Fundamentals
Contents
Fundamentals#
In the sections below, key components of the BioNeMo framework and their use will be discussed
Overview & Core Features#
The NVIDIA BioNeMo framework exists for training and deploying large biomolecular language models at supercomputing scale for the discovery and development of therapeutics. The large language model (LLM) framework currently has models for small molecules (SMILES) and protein sequences. Its modular design and high-level APIs make it easy to create, train, and deploy complex models for a variety of downstream tasks. The extensive collection of pre-trained models and scripts further facilitates rapid prototyping and development, and developers can customize the repository according to their needs.
Note
For detailed information on model pre-training or fine-tuning, users should consult the Tutorials section. This BioNeMo Core section is intended for advanced use cases.
Creating New Models#
BioNeMo models implement classes that inherit from NeMo Collections. For example, ESM-1nv inherits functions from NeMo’s Megatron Bert Model.
class ESM1nvModel(MegatronBertModel):
"""
ESM1nv pre-training
"""
def __init__(self, cfg: DictConfig, trainer: Trainer):
super().__init__(cfg, trainer=trainer)
(...)
Re-using code and architectures is the easiest way to modify existing BioNeMo pipelines. Creation of a new model in BioNeMo can be accomplished by leveraging NeMo Collections as a base model from which functions can be inherited. Similarly, existing BioNeMo implementations can be modified to create additional downstream tasks. This is the most straight-forward way to add additional building blocks to the repository. For example, in the snippet below found in bionemo/model/protein/esm1nv/esm1nv_model.py the tokenization method can be customized from the ESM-1nv implementation
def _build_tokenizer(self):
"""
Default tokenizer is based on available nemo tokenizers.
Override this method to use an external tokenizer.
All tokenizers are expected to provide compatible interface.
"""
self.tokenizer = get_nmt_tokenizer(
library=self._cfg.tokenizer.library,
tokenizer_model=self.register_artifact("tokenizer.model", self._cfg.tokenizer.model),
vocab_file=self.register_artifact("tokenizer.vocab_file", self._cfg.tokenizer.vocab_file),
legacy=False,
)
The same customization principles apply to all the other models available in BioNeMo framework, but certain nuances apply to each one of them according to their super class. MegaMolBART, for example, does not have the same @property decorated functions dealing with input and output names or input types as ESM1-nv:
# ESM1-nv
@property
def input_types(self) -> Optional[Dict[str]]:
return {
'input_ids': {
0: 'batch',
1: 'time'
},
'attention_mask': {
0: 'batch',
1: 'time'
}
}
@property
def output_types(self) -> Optional[Dict[str]]:
return {
'output': {
0: 'batch',
1: 'time',
2: 'size'
}
}
On the other hand, the ESM-1nv implementation, which is based on a BERT (an encoder model), cannot sample and decode molecules as is done by MegaMolBART, which is based on BART (an encoder-decoder model).
# MegaMolBART
def sample_molecules(self, tokens_enc, enc_mask, hidden_states=None):
(...)
self.freeze()
# Decode encoder hidden state to tokens
predicted_tokens_ids, log_probs = self.decode(tokens_enc,
enc_mask,
self._cfg.max_position_embeddings,
enc_output=hidden_states)
predicted_tokens_ids = predicted_tokens_ids.cpu().detach().numpy().tolist()
# Prune tokens by eos / padding and convert to SMILES
for item, predicted_tokens_ in enumerate(predicted_tokens_ids):
if self.tokenizer.eos_id in predicted_tokens_:
idx = predicted_tokens_.index(self.tokenizer.eos_id)
predicted_tokens_ids[item] = predicted_tokens_[:idx]
else:
# NB: this is slightly different from previous version in that pad tokens can be in the middle of sequence
predicted_tokens_ids[item] = [id for id in predicted_tokens_ if id != self.tokenizer.pad_id]
predicted_tokens_text = self.tokenizer.ids_to_tokens(predicted_tokens_ids)
sampled_smiles = self.tokenizer.tokens_to_text(predicted_tokens_text)
self.unfreeze()
return sampled_smiles
Here’s a summary of each model’s superclass:
Model |
Superclass |
|---|---|
ESM-1nv |
MegatronBertModel |
ProtT5nv |
MegatronT5Model |
MegaMolBART |
MegatronLMEncoderDecoderModel |
Customizations can also vary from simple changes in the tokenizer methods to more involved changes in the model architecture, including alterations in how the forward step is computed, callbacks or data augmentation functions.
Config Files#
BioNeMo framework offers the option to easily set up and change model configurations for pre-training or fine-tuning workflows. Such configuration modifications can range from simple hyperparameters, such as number of hidden states to more advanced oodifications for data handling or connections to Weights & Biases Dashboards for experiment tracking; for example, refer to bionemo/examples/protein/esm1nv/conf/base_config.yaml
Examples of typical configuration modifications for training can be fouind in the .yaml files within the framework
trainer:
devices: 8 # number of GPUs
num_nodes: 2 # If you are working with a multi-node setting, for example, 2xDGX systems
precision: 16 # FP 16, 32
(...)
accumulate_grad_batches: 1
BioNeMo also makes it easier to distribute weights for models that are too large for a single GPU onto multiple GPUs. A simple change in the tensor_model_parallel_size will make BioNeMo distribute a model’s layers across GPUs to better manage memory loads as weights are computed.
model:
micro_batch_size: 1
global_batch_size: 2
tensor_model_parallel_size: 1 # Increase this number to distribute the model across GPUs
(...)
seq_length: 512
(...)
num_attention_heads: 12
activation: 'gelu' # Options ['gelu', 'geglu', 'swiglu', 'reglu']
(...)
It is also straightforward to change the configuration file to download and process datasets:
# Path to data must be specified by the user.
data_url: 'https://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref50/uniref50.fasta.gz'
dataset_path: /data/uniref2022_05 # parent directory for data, contains train / val / test folders
dataset: # inclusive range of data files to load or can load a single file, for example, x000.csv
train: x000
test: x000
val: x000
(...)
seq_length_dec: 256 # Target sequence length
skip_warmup: True
num_workers: 16 # number of workers to be used for dataset preprocessing. 0 -- all available workers
dataloader_type: single
Some configuration parameters are inter-dependent. For example, global_batch_size, if provided, must be computed by a formula micro_batch_size * devices * accumulate_grad_batches / (tensor_model_parallel_size * pipeline_model_parallel_size). For simplicity, global_batch_size can be left to null, and the appropriate value will be inferred automatically.
There are also configuration parameters that are relevant to the model’s tasks and multiple config files can be set. For example, under examples/molecule/megamolbart/ there are several config files, each of which establishing a specific behavior for pre-training tasks.
These configuration parameters should be reviewed prior to the first run to minimize chances of errors such as CUDA Out of Memory or low levels of GPU utilization in multi-node settings. For more information about configuration parameters check the files provided in the conf folder. To get more details about typical issues, refer to the Frequently Asked Questions or to the NVIDIA Developer Forums.
One additional feature of note is that ranges of data files can be selected, if the data are sharded into multiple files. For example, to select a contiguous range of 100 data files, from x000.csv to x099.csv, use the range indicator x[000..099]. For only ten files, use x[000..009]. Ensure these are set as appropriate for the train, validation, and test splits as below in the YAML config file:
model:
data:
dataset:
train: x[000..099]
test: x[000..099]
val: x[000..099]
Command Line Configuration#
BioNeMo framework users can count on resources improved management of containers and configuration settings of complex applications. There are two main components employed for that: NGC CLI and Hydra.
NVIDIA NGC CLI is a command-line interface tool for managing Docker containers in the NVIDIA NGC Registry. With NGC CLI, you can perform the same operations that are available from the NGC website, such as running jobs, viewing ACE and node information, and viewing Docker repositories within your orgs. For more information about how to set up CLI, check the NGC CLI SETUP page and NGC CLI Docs.
Hydra is a framework for simplifying the configuration of complex applications and environments. In the BioNeMo framework, the most common application of Hydra is to handle YAML-based configuration files and their parameters. YAML is a human-readable data serialization standard that can be used in conjunction with all programming languages and is often used to write configuration files.
For example, to define training task using ESM-1nv, one could start the main method with a @hydra runner decorator, defined by the creators of the NeMo Framework more details on GitHub. This decorator is used for passing the config paths to main function and optionally registers a schema used for validation/providing default values.
@hydra_runner(config_path="<configuration file path>", config_name="<name of the configuration>")
def main(cfg) -> None:
(...)
trainer = setup_trainer(cfg)
if cfg.do_training:
model = ESM1nvModel(cfg.model, trainer)
trainer.fit(model)
(...)
The @hydra_runner Decorator can also help in multi-run configs. This can be useful for hyperparameter search and other tasks involving experimentation. To check detailed instructions about using @hydra_runner check the NeMo Experiment Manager documentation.
Typically, the config files can be found under the conf directory in the same folder as the training script. For example, the config file for pre-training a ProtT5 model is located at examples/protein/prott5nv/conf/pretrain_small.yaml.
Checkpoints#
Pre-trained checkpoints are also provided based on the models described in the Introduction. These checkpoints will have token-size limitations.
Model |
Checkpoint’s Max. Length |
Common Tasks |
|---|---|---|
ESM-1nv & ProtT5nv |
512 Tokens (Amino Acids) |
Thermostability, Secondary Structure Prediction, Subcellular Localization (check FLIP Benchmark) |
MegaMolBART |
512 Tokens (SMILES) |
Representation Learning, Structure Prediction, Molecule Generation |
To enable support for longer sequences, customize configuration parameters to allow sequence lengths, assuming the user has the computational resources to support expansion. Taking ProtT5nv as example, several different configuration options are available in the .yaml file. Under examples/protein/prott5nv/conf/ the base_config.yaml file is the basis for production-level work. More information about how to use checkpoints can be found in the Quickstart Guide and in the Save-Restore Connectors section below.
Save-Restore Connectors#
The Save-Restore Connector (BioNeMoSaveRestoreConnector) is a component of BioNeMo framework. This class inherits functions from NLPSaveRestoreConnector from the NeMo Framework. These connectors are designed to handle the loading and modification of model checkpoints, for example, so that they can support changes in vocabulary size or a different type of positional embeddings. A Save-Restore connector object is supplied when initializing model from an existing checkpoint via the model restore_from method.
The restore_from_path config parameter can be added to .yaml files as one of the cutomizations mentioned before. For example, under examples/conf/ the base_infer_config.yaml file has the following parameters:
model:
downstream_task:
restore_from_path: ??? # path of pretrained model to be used in inference
Which is invoked in several occasions, including in the pretrain.py file for the ProtT5nv model
if cfg.do_training:
logging.info("************** Starting Training ***********")
if cfg.restore_from_path is not None:
logging.info("\nRestoring model from .nemo file " + cfg.restore_from_path)
model = ProtT5nvModel.restore_from(
cfg.restore_from_path, cfg.model, trainer=trainer,
save_restore_connector=BioNeMoSaveRestoreConnector(vocab_size=128),
strict=False
)
Modifying a Checkpoint Dictionary#
The save-restore connector is useful in situations when modifying the dictionary for an existing checkpoint, either to accommodate fewer or more tokens than used for the previous training. This allows flexibility when fine-tuning models with proprietary datasets.
Fine Tuning Pre-trained Checkpoints#
Given the ease to operate with Data Module and Save-Restore Connectors, The BioNeMo Framework also makes it simple to organize and use checkpoints. A demo for implementing the EncoderFineTuning class from scratch, adding data, modifying the configuration parameters, and creating a fine tuning script can be found here.
Datasets#
BioNeMo framework pipelines are configured for specific dataset formats.
Model |
Data Type |
Common Public Datasets |
|---|---|---|
ESM-1nv & ProtT5nv |
Proteins (FASTA, FASTQ, PDB) |
UniProt, GenBank, SRA, PDB, |
MegaMolBART |
Molecules (CSV or SMI) |
PubChem, ZINC, ChEMBL, DUD-E |
However, the pipelines can be modified for different datasets. The following three tutorials describe how to modify the ESM-1nv pipeline for compatibility with the Observed Antibody Space (OAS) database:
[Creating a Custom Dataloader](TUTORIAL LINK)
Weights & Biases Integration#
API Setup#
Enabling integration with Weights & Biases is highly recommended. To leverage this feature, ensure the .env file contains a WANDB API key entered for the variable WANDB_API_KEY. Check the Weights and Biases user guide for further instructions on obtaining an API key. Refer to the Quickstart Guide for details on setup.
Weights and Biases Charts#
In the image below are examples of metrics and dashboards used to monitor model training progress.
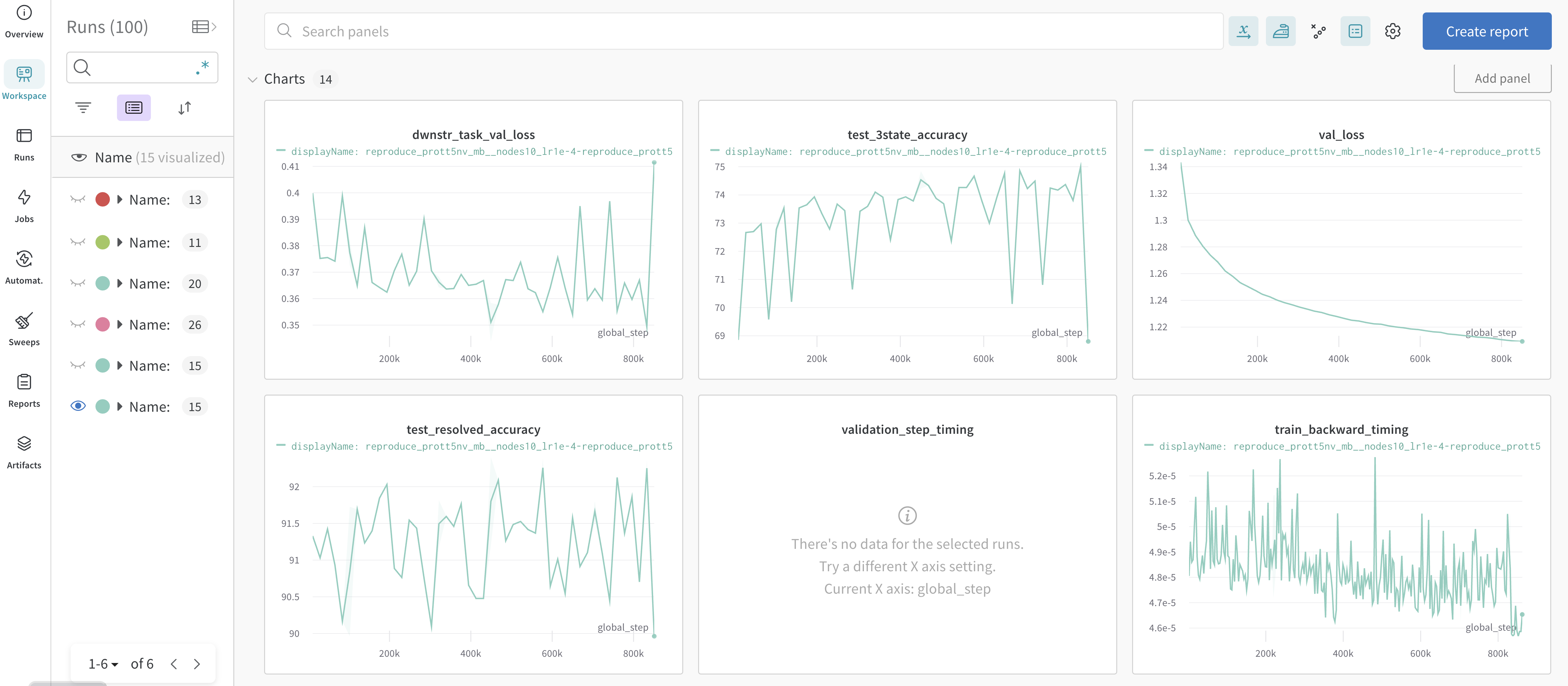
These training and system related metrics are automatically logged in the online dashboard when an API key is provided and upload to Weights and Biases is enabled.
Training related metrics include:
Reduced Train Loss: is the value of the training loss function aggregated from all parallel processes. If the training loss doesn’t decrease or explodes, this is a possible sign that the learning rate needs to be reduced.
Loss Scale: the scaling factor of the loss.
Gradient Norm: the value of the gradient norm. Increasing or undefined (NaN) values of the gradient norm usually indicate instabilities in training. In such cases, the learning rate may need to be reduced.
Learning Rate: the value of the learning rate.
Epoch: the value of the epoch. Note: Megatron datasets upsample the data, so the entire training up to
max_stepsis considered a single epoch.Consumed Samples: the number of training samples that have been consumed during training.
Validation Step Timing: monitors the time required for each validation step during training. this is useful for diagnosing performance issues and bottlenecks in the validation process, and can help optimize the speed and efficiency of model training.
Train Backward Timing: a measure of the time required for the backpropagation step. Measuring the time this takes can help identify bottlenecks in the training process and assist in performance optimization.
Validation Loss: the loss function computed on the validation set. Validation loss is used during model training to avoid over-fitting (when a model learns the training data too well and performs poorly on unseen data). If validation loss starts to increase while training loss decreases, this is usually a sign of over-fitting.
Refer to also optional metrics for Validation With a Downstream Task