Train MolMIM from scratch on your own data using the BioNeMo Framework
Contents
Train MolMIM from scratch on your own data using the BioNeMo Framework#
The purpose of this tutorial is to provide an example use case of training MolMIM model using the BioNeMo framework.
Demo objectives:#
Learn how to prepare your own train, validation and test datasets for MolMIM training -> data processing steps including input validation, deduplication, filtering based on tokenizer vocabulary, dataset splitting
Train a MolMIM model from scratch (highlighting config options for customisable training)
Continue training an existing MolMIM model checkpoint
Note: this notebook was developed and tested for BioNeMo framework container 1.7
Tested GPUs: A1000, A6000 (total notebook runtime using single GPU ~2 mins)
Overview - MolMIM model#
MolMIM is a probabilistic auto-encoder for small molecule drug discovery, trained with Mutual Information Machine (MIM) learning. It provides a fixed size embedding for each molecule and produces a latent space from which samples can be drawn and novel SMILES strings generated from a specific starting molecule. By using optimizers in tandem with MolMIM, specific properties of the molecules can be optimized and novel derivatives generated. For more information, we direct the reader to Reidenbach, et al. (2023).
Architecture (see schematic below):
Perceiver Encoder
Transformer Decoder
Latent MLP heads for latent variable characterization
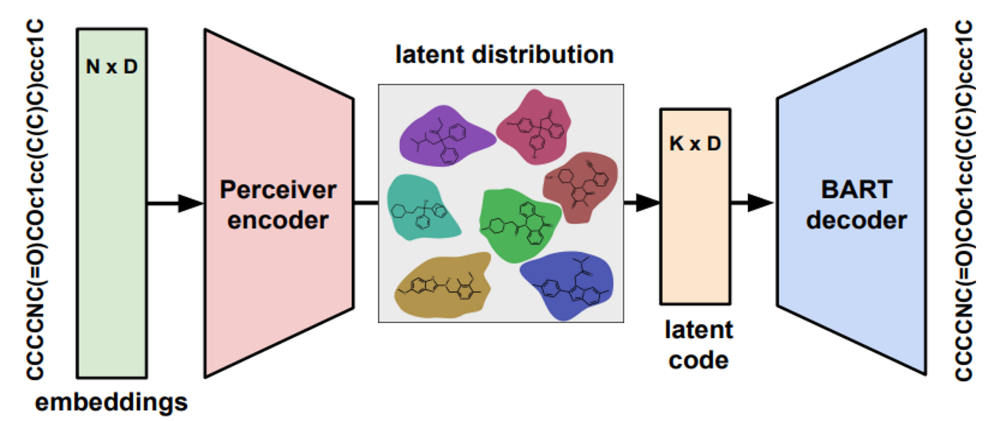
Current MolMIM models were pretrained using only molecules that conform to Lipinski’s rule of 5, here we will give an example of how you could train a custom model on molecules of your choice, without filtering using the Rule of 5.
Setup and Assumptions#
This tutorial assumes that the user has access to BioNeMo framework container and GPU compute, please check the Getting Started section for more information.
All model training related commands should be executed inside the BioNeMo docker container.
Note: The interactive job launch example shown here using the Jupyter Lab interface is intended for initial user experience/trial runs. It is strongly advised to launch the model training jobs using the launch script as a part of the ngc batch run command, as mentioned in Access and Startup.
%%capture --no-display --no-stderr cell_outputto suppress this output. Comment or delete this line in the cells below to restore full output.
First we install and import all our required packages
%%capture --no-display --no-stderr cell_output
! pip install PyTDC
# Importing required libraries
import numpy as np
import os
from pathlib import Path
import pandas as pd
from rdkit import Chem
from typing import Literal
import warnings
warnings.filterwarnings('ignore')
warnings.simplefilter('ignore')
# Importing libraries to download and split datasets from the Therapeutic Data Commons https://tdcommons.ai/
from tdc.single_pred import Tox
from tdc.base_dataset import DataLoader
from nemo.collections.common.tokenizers.regex_tokenizer import RegExTokenizer
bionemo_home = os.environ['BIONEMO_HOME']
1. Dataset preparation#
Here we go through data preparation steps for an example dataset from the Therapeutic Data Commons: a regression dataset - the percent inhibition of the human ether-à-go-go related gene (hERG) channel by a given compound at a 10 µM concentration. This dataset contains a total of 306892 molecules.
Download the dataset#
First we download the dataset, and perform some basic data processing, for example canonicalising the SMILES strings and dropping duplicate entries.
def canonicalise_smiles(smiles: str) -> str:
"""Returns the canonical SMILES string for the input SMILES string
or np.nan if it was not possible to generate a valid mol object from the input string
"""
mol = Chem.MolFromSmiles(smiles)
return np.nan if mol is None else Chem.MolToSmiles(mol)
# Specify dataset for download
dataset_name = 'herg_central'
task = 'hERG_at_10uM'
print(f"Preparing raw {dataset_name} dataset from TD Commons, filtered for task: {task}.")
data = Tox(name = dataset_name, label_name = task)
data_df = data.get_data()
# Take only the first 10,000 molecules in order to reduce tutorial run time
data_df = data_df[:10000]
print("Generating canonical SMILES strings from the provided SMILES...")
data_df["canonical_smiles"] = data_df["Drug"].map(canonicalise_smiles)
print("Dropping duplicate molecules (first instance kept)...")
unique_df = data_df.drop_duplicates(subset=["canonical_smiles"])
print(f"{len(data_df) - len(unique_df)} duplicates removed.")
Found local copy...
Loading...
Preparing raw herg_central dataset from TD Commons, filtered for task: hERG_at_10uM.
Done!
Generating canonical SMILES strings from the provided SMILES...
Dropping duplicate molecules (first instance kept)...
0 duplicates removed.
Filter for vocabulary compliance#
Next, we exclude SMILES strings exceeding the model’s maximum token limit and molecules with tokens absent from the model’s vocabulary. To accomplish this, we import the model’s existing vocabulary files.
max_token_length = 126
# Note: the maximum token length generated from the smiles string should be 2 less than the max_seq_length specified in the model config.
# This is to account for the extra tokens <BOS> and <EOS>
def vocab_compliance_check(smiles: str, tokenizer: RegExTokenizer, max_token_length: int) -> bool:
"""Checks if the SMILES string only contains vocabulary in the tokenizer's vocabulary
and if the token length is less than or equal to `max_token_length"""
tokens = tokenizer.text_to_tokens(smiles)
vocab_allowed = tokenizer.vocab.keys()
return set(tokens).issubset(set(vocab_allowed)) and len(tokens) <= max_token_length
model_name = "molmim"
print(f"Filtering out molecules which are not present in the {model_name} tokenizer vocabulary or with max token length greater than {max_token_length}...")
tokenizer_path = bionemo_home + "/tokenizers/molecule/{model_name}/vocab/{model_name}.{extension}"
tokenizer = RegExTokenizer().load_tokenizer(regex_file=tokenizer_path.format(model_name=model_name, extension="model"), vocab_file=tokenizer_path.format(model_name=model_name, extension="vocab"))
unique_df["vocab_compliant"] = unique_df["canonical_smiles"].apply(lambda smi: vocab_compliance_check(smi, tokenizer, max_token_length))
# Select only molecules which are vocab compliant
filtered_df= unique_df.loc[unique_df['vocab_compliant']]
print(f"{len(unique_df) - len(filtered_df)} molecules removed.")
Filtering out molecules which are not present in the molmim tokenizer vocabulary or with max token length greater than 126...
[NeMo I 2024-09-03 15:07:43 regex_tokenizer:240] Loading vocabulary from file = /workspace/bionemo/tokenizers/molecule/molmim/vocab/molmim.vocab
[NeMo I 2024-09-03 15:07:43 regex_tokenizer:254] Loading regex from file = /workspace/bionemo/tokenizers/molecule/molmim/vocab/molmim.model
1 molecules removed.
Split dataset to create training, validation and test sets#
Finally, we split the dataset into training, validation, and test sets with a ratio of 7:1:2, respectively. These sets are then saved as CSV files in designated subdirectories (train, val and test) as required for model training. Other dataset variables e.g. columns, headers, dataset_path are specified in the config used for training, see example config files in examples/conf/molecule for more information.
We use a scaffold split here to make the test set more difficult, as it contains molecules with different structures.
def generate_splits(data: DataLoader, filtered_df: pd.DataFrame, method: Literal['scaffold', 'random'], seed: int, frac: list) -> dict:
"""Splits the data into train, validation and test sets by the specified method, according to the specified fractions
and returns a dictionary of dataframes
"""
# Update data object with filtered molecules
data.entity1 = filtered_df.canonical_smiles
data.entity1_idx = filtered_df.Drug_ID
data.y = filtered_df.Y
print(f"Generating train/valid/test sets according to {method} strategy in the following fractions of the total dataset: {frac}")
return data.get_split(method=method, seed=seed, frac=frac)
splits = generate_splits(data, filtered_df, method='scaffold', seed=42, frac= [0.7,0.1,0.2])
directory_mapping = {"valid":"val"}
print("Writing sets to file...")
for molecule_set in splits.keys():
set_name = directory_mapping.get(molecule_set, molecule_set)
splits[molecule_set].rename(columns= {"Drug":"smiles"}, inplace =True)
output_dir = os.path.join(bionemo_home, f"data/processed/{task}/{set_name}")
Path(output_dir).mkdir(parents=True, exist_ok=True)
outfilename = os.path.join(output_dir, f"{set_name}_set.csv")
splits[molecule_set].to_csv(outfilename, index=False)
print(f"Saved {set_name} set molecules to {outfilename}")
Generating train/valid/test sets according to scaffold strategy in the following fractions of the total dataset: [0.7, 0.1, 0.2]
100%|█████████████████████████████████████████████████████████████████████████████████| 9999/9999 [00:01<00:00, 6815.68it/s]
Writing sets to file...
Saved train set molecules to /workspace/bionemo/data/processed/hERG_at_10uM/train/train_set.csv
Saved val set molecules to /workspace/bionemo/data/processed/hERG_at_10uM/val/val_set.csv
Saved test set molecules to /workspace/bionemo/data/processed/hERG_at_10uM/test/test_set.csv
2. Pretrain MolMIM from scratch#
Here we will run pretraining from scratch using the default config “pretrain_xsmall_canonicalized.yaml”. Other example configs can be found in directory examples/molecule/molmim/conf. We will override some config arguments at runtime using Hydra.
The column containing the SMILES strings in the input datasets is specified with the argument model.data.data_impl_kwargs.csv_mmap.data_col. We specify the directory containing our newly created train, val and test subdirectories using the argument model.data.dataset_path and the names of our input files using argument model.data.dataset.train etc.
Note, index files are written to the file specified in model.data.index_mapping_dir, if index files here are present they will be read rather than written. Therefore, we here clear the index files first to avoid unintentional errors which could occur if changing model or dataset params in this notebook playground.
For the same reason we set exp_manager.resume_if_exists to be False, otherwise if training was interrupted and then restarted, training will continue from the last saved checkpoint, and errors could occur if model params in the config had been changed.
We will also reduce the number of training steps (trainer.max_steps) and correspondingly the step interval for checking the validation set (trainer.val_check_interval) just for the purpose of this demonstration.
Optional: Add arguments for logging with Weights and Biases and login when prompted
%%capture --no-display --no-stderr cell_output
! rm -rf data/data_index
! cd {bionemo_home} && python examples/molecule/molmim/pretrain.py \
do_training=True \
++model.data.dataset_path="data/processed/{task}/" \
++model.data.dataset.train="train_set" \
++model.data.dataset.val="val_set" \
++model.data.dataset.test="test_set" \
++model.data.index_mapping_dir="data/data_index/" \
++model.data.data_impl_kwargs.csv_mmap.data_col=1 \
++model.dwnstr_task_validation.enabled=False \
++model.global_batch_size=null \
++trainer.devices=1 \
++trainer.accelerator='gpu' \
++trainer.max_steps=200 \
++trainer.val_check_interval=100 \
++exp_manager.create_wandb_logger=False \
++exp_manager.resume_if_exists=False
3. Continue training on an existing model checkpoint#
To do this, we run the pretrain.py script but specify the ++exp_manager.resume_if_exists=True argument. We will use the model we just trained above and we will override some config arguments at runtime using Hydra. Specifically, we will increase the max_steps argument to 400, which will train an additional 200 steps on top of the 200 that were taken in the initial training run.
Note: ensure the config specified matches the existing model to be loaded.
%%capture --no-display --no-stderr cell_output
model_path = "/result/nemo_experiments/MolMIM/MolMIM-xsmall_pretraining/checkpoints/MolMIM.nemo"
! rm -rf data/data_index
! cd {bionemo_home} && python examples/molecule/molmim/pretrain.py \
do_training=True \
++model.data.dataset_path="data/processed/{task}/" \
++model.data.dataset.train="train_set" \
++model.data.dataset.val="val_set" \
++model.data.dataset.test="test_set" \
++model.data.index_mapping_dir="data/data_index/" \
++model.data.data_impl_kwargs.csv_mmap.data_col=1 \
++model.dwnstr_task_validation.enabled=False \
++model.global_batch_size=null \
++trainer.devices=1 \
++trainer.accelerator='gpu' \
++trainer.max_steps=400 \
++trainer.val_check_interval=100 \
++exp_manager.create_wandb_logger=False \
++exp_manager.resume_if_exists=True