deepvariant_germline
Given one or more pairs of FASTQ files, you can run the germline variant tool to generate BAM, variants, duplicate metrics and recal.
The deepvariant germline tool includes alignment, sorting, and marking as well as the DeepVariant variant caller.
The inputs are BWA-indexed reference files and pair-ended FASTQ files. The outputs of this tool are the following:
Aligned, co-ordinate sorted, duplicated marked BAM
Variants in
vcf/g.vcf/g.vcf.gzformat
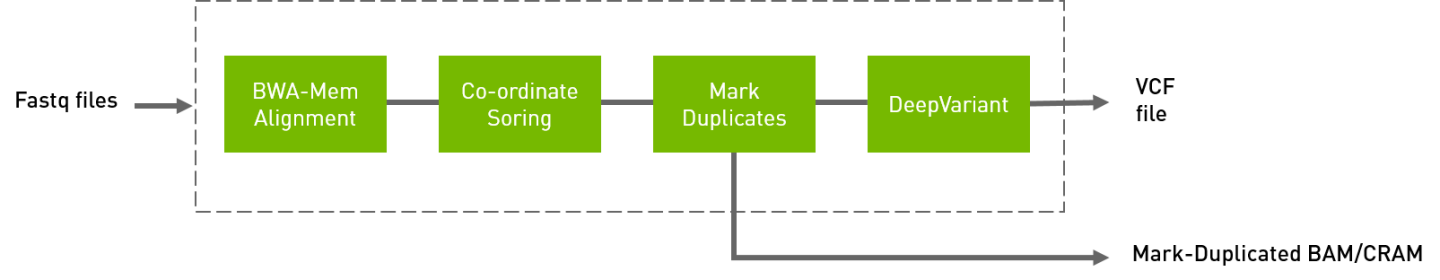
The following command runs the DeepVariant tool.
# This command assumes all the inputs are in INPUT_DIR and all the outputs go to OUTPUT_DIR.
docker run --rm --gpus all --volume INPUT_DIR:/workdir --volume OUTPUT_DIR:/outputdir \
--workdir /workdir \
nvcr.io/nvidia/clara/clara-parabricks:4.2.1-1 \
pbrun deepvariant_germline \
--ref /workdir/${REFERENCE_FILE} \
--in-fq /workdir/${INPUT_FASTQ_1} /workdir/${INPUT_FASTQ_2} \
--out-variants /outputdir/${OUTPUT_VCF_FILE}
The commands below are the Google counterpart of the Parabricks command above. The output from these commands will be identical to the output from the above command. See the Output Comparison page for comparing the results.
# Run bwa-mem and pipe output to create sorted BAM
$ bwa mem \
-t 32 \
-K 10000000 \
-R '@RG\tID:sample_rg1\tLB:lib1\tPL:bar\tSM:sample\tPU:sample_rg1' \
<INPUT_DIR>/${REFERENCE_FILE} \
<INPUT_DIR>/${INPUT_FASTQ_1} <INPUT_DIR>/${INPUT_FASTQ_2} | \
gatk SortSam \
--java-options -Xmx30g \
--MAX_RECORDS_IN_RAM 5000000 \
-I /dev/stdin \
-O cpu.bam \
--SORT_ORDER coordinate
# Mark Duplicates
$ gatk MarkDuplicates \
--java-options -Xmx30g \
-I cpu.bam \
-O mark_dups_cpu.bam \
-M metrics.txt
# Run deepvariant
BIN_VERSION="1.5.0"
sudo docker run \
-v "${PWD}":"/input" \
-v "${PWD}/output":"/output" \
-v "${PWD}/Ref":"/reference" \
google/deepvariant:"${BIN_VERSION}" \
/opt/deepvariant/bin/run_deepvariant \
--model_type WGS \
--ref /reference/Homo_sapiens_assembly38.fasta \
--reads /output/mark_dups_cpu.bam \
--output_vcf /output/"${OUTPUT_VCF_FILE}" \
--num_shards $(nproc) \
--make_examples_extra_args "ws_use_window_selector_model=true"
See the DeepVariant Models for additional GPUs section for instructions on downloading and using model files for additional GPUs.
Run the germline pipeline from FASTQ to VCF using a deep neural network analysis.
Input/Output file options
- --ref REF
- --in-fq [IN_FQ [IN_FQ ...]]
- --in-se-fq [IN_SE_FQ [IN_SE_FQ ...]]
- --knownSites KNOWNSITES
- --interval-file INTERVAL_FILE
- --pb-model-file PB_MODEL_FILE
- --out-recal-file OUT_RECAL_FILE
- --out-bam OUT_BAM
- --out-variants OUT_VARIANTS
- --out-duplicate-metrics OUT_DUPLICATE_METRICS
- --proposed-variants PROPOSED_VARIANTS
Path to the reference file. (default: None)
Option is required.
Path to the pair-ended FASTQ files followed by optional read groups with quotes (Example: "@RG\tID:foo\tLB:lib1\tPL:bar\tSM:sample\tPU:foo"). The files must be in fastq or fastq.gz format. All sets of inputs should have a read group; otherwise, none should have a read group, and it will be automatically added by the pipeline. This option can be repeated multiple times. Example 1: --in-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz --in-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz. Example 2: --in-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz "@RG\tID:foo\tLB:lib1\tPL:bar\tSM:sample\tPU:unit1" --in-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz "@RG\tID:foo2\tLB:lib1\tPL:bar\tSM:sample\tPU:unit2". For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU. (default: None)
Path to the single-ended FASTQ file followed by optional read group with quotes (Example: "@RG\tID:foo\tLB:lib1\tPL:bar\tSM:sample\tPU:foo"). The file must be in fastq or fastq.gz format. Either all sets of inputs have a read group, or none should have one, and it will be automatically added by the pipeline. This option can be repeated multiple times. Example 1: --in-se-fq sampleX_1.fastq.gz --in-se-fq sampleX_2.fastq.gz . Example 2: --in-se-fq sampleX_1.fastq.gz "@RG\tID:foo\tLB:lib1\tPL:bar\tSM:sample\tPU:unit1" --in-se-fq sampleX_2.fastq.gz "@RG\tID:foo2\tLB:lib1\tPL:bar\tSM:sample\tPU:unit2" . For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU. (default: None)
Path to a known indels file. The file must be in vcf.gz format. This option can be used multiple times. (default: None)
Path to an interval file in one of these formats: Picard-style (.interval_list or .picard), GATK-style (.list or .intervals), or BED file (.bed). This option can be used multiple times. (default: None)
Path to a non-default parabricks model file for deepvariant. (default: None)
Path of the report file after Base Quality Score Recalibration. (default: None)
Path of BAM file after Marking Duplicates. (default: None)
Option is required.
Path of the vcf/gvcf/gvcf.gz file after variant calling. (default: None)
Option is required.
Path of a duplicate metrics file after Marking Duplicates. (default: None)
Path of the VCF file, which has proposed variants for the make examples stage. (default: None)
Tool Options:
- -L INTERVAL, --interval INTERVAL
- --bwa-options BWA_OPTIONS
- --no-warnings
- --filter-flag FILTER_FLAG
- --skip-multiple-hits
- --min-read-length MIN_READ_LENGTH
- --align-only
- --no-markdups
- --fix-mate
- --markdups-assume-sortorder-queryname
- --markdups-picard-version-2182
- --optical-duplicate-pixel-distance OPTICAL_DUPLICATE_PIXEL_DISTANCE
- --read-group-sm READ_GROUP_SM
- --read-group-lb READ_GROUP_LB
- --read-group-pl READ_GROUP_PL
- --read-group-id-prefix READ_GROUP_ID_PREFIX
- --standalone-bqsr
- --max-read-length-fq2bamfast MAX_READ_LENGTH_FQ2BAMFAST
- --min-read-length-fq2bamfast MIN_READ_LENGTH_FQ2BAMFAST
- --disable-use-window-selector-model
- --gvcf
- --norealign-reads
- --sort-by-haplotypes
- --keep-duplicates
- --vsc-min-count-snps VSC_MIN_COUNT_SNPS
- --vsc-min-count-indels VSC_MIN_COUNT_INDELS
- --vsc-min-fraction-snps VSC_MIN_FRACTION_SNPS
- --vsc-min-fraction-indels VSC_MIN_FRACTION_INDELS
- --min-mapping-quality MIN_MAPPING_QUALITY
- --min-base-quality MIN_BASE_QUALITY
- --mode MODE
- --alt-aligned-pileup ALT_ALIGNED_PILEUP
- --variant-caller VARIANT_CALLER
- --add-hp-channel
- --parse-sam-aux-fields
- --use-wes-model
- --include-med-dp
- --normalize-reads
- --pileup-image-width PILEUP_IMAGE_WIDTH
- --channel-insert-size
- --no-channel-insert-size
- --max-read-size-512
- --prealign-helper-thread
- --track-ref-reads
- --phase-reads
- --dbg-min-base-quality DBG_MIN_BASE_QUALITY
- --ws-min-windows-distance WS_MIN_WINDOWS_DISTANCE
- --channel-gc-content
- --channel-hmer-deletion-quality
- --channel-hmer-insertion-quality
- --channel-non-hmer-insertion-quality
- --skip-bq-channel
- --aux-fields-to-keep AUX_FIELDS_TO_KEEP
- --vsc-min-fraction-hmer-indels VSC_MIN_FRACTION_HMER_INDELS
- --vsc-turn-on-non-hmer-ins-proxy-support
- --consider-strand-bias
- --p-error P_ERROR
- --channel-ins-size
- --max-ins-size MAX_INS_SIZE
- --disable-group-variants
- --filter-reads-too-long
Interval within which to call bqsr from the input reads. All intervals will have a padding of 100 to get read records, and overlapping intervals will be combined. Interval files should be passed using the --interval-file option. This option can be used multiple times e.g. "-L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000". (default: None)
Pass supported bwa mem options as one string. The current original bwa mem supported options are -M, -Y and -T e.g. --bwa-options="-M -Y" (default: None)
Suppress warning messages about system thread and memory usage. (default: None)
Don't generate SAM entries in the output if the entry's flag's meet this criteria. Criteria: (flag & filter != 0) (default: 0)
Filter SAM entries whose length of SA is not 0. (default: None)
Skip reads below minimum read length. They will not be part of the output. (default: None)
Generate output BAM after bwa-mem. The output will not be co-ordinate sorted or duplicates will not be marked. (default: None)
Do not perform the Mark Duplicates step. Return BAM after sorting. (default: None)
Add mate cigar (MC) and mate quality (MQ) tags to the output file. (default: None)
Assume the reads are sorted by queryname for Marking Duplicates. This will mark secondary, supplementary, and unmapped reads as duplicates as well. This flag will not impact variant calling while increasing processing times. (default: None)
Assume marking duplicates to be similar to Picard version 2.18.2. (default: None)
The maximum offset between two duplicate clusters in order to consider them optical duplicates. Ignored if --out-duplicate-metrics is not passed. (default: None)
SM tag for read groups in this run. (default: None)
LB tag for read groups in this run. (default: None)
PL tag for read groups in this run. (default: None)
Prefix for the ID and PU tags for read groups in this run. This prefix will be used for all pairs of fastq files in this run. The ID and PU tags will consist of this prefix and an identifier, that will be unique for a pair of fastq files. (default: None)
Run standalone BQSR. (default: None)
Maximum read length/size (i.e., sequence length) used for bwa and filtering FASTQ input (Argument only applies to --fq2bamfast) (default: 480)
Minimum read length/size (i.e., sequence length) used for bwa and filtering FASTQ input (Argument only applies to --fq2bamfast) (default: 10)
Change the window selector model from Allele Count Linear to Variant Reads. This option will increase the accuracy and runtime. (default: None)
Generate variant calls in .gvcf Format. (default: None)
Do not locally realign reads before calling variants. Reads longer than 500 bp are never realigned. (default: None)
Reads are sorted by haplotypes (using HP tag). (default: None)
Keep reads that are duplicate. (default: None)
SNP alleles occurring at least this many times in the AlleleCount will be advanced as candidates. (default: 2)
Indel alleles occurring at least this many times in the AlleleCount will be advanced as candidates. (default: 2)
SNP alleles occurring at least this fraction of all counts in the AlleleCount will be advanced as candidates. (default: 0.12)
Indel alleles occurring at least this fraction of all counts in the AlleleCount will be advanced as candidates. (default: None)
By default, reads with any mapping quality are kept. Setting this field to a positive integer i will only keep reads that have a MAPQ >= i. Note this only applies to aligned reads. (default: 5)
Minimum base quality. This option enforces a minimum base quality score for alternate alleles. Alternate alleles will only be considered if all bases in the allele have a quality greater than min_base_quality. (default: 10)
Value can be one of [shortread, pacbio, ont]. By default, it is shortread. If mode is set to pacbio, the following defaults are used: --norealign-reads, --alt-aligned-pileup diff_channels, --vsc-min-fraction-indels 0.12. If mode is set to ont, the following defaults are used: -norealign-reads, --variant-caller VCF_CANDIDATE_IMPORTER. (default: shortread)
Value can be one of [none, diff_channels]. Include alignments of reads against each candidate alternate allele in the pileup image. (default: None)
Value can be one of [VERY_SENSITIVE_CALLER, VCF_CANDIDATE_IMPORTER]. The caller to use to make examples. If you use VCF_CANDIDATE_IMPORTER, it implies force calling. Default is VERY_SENSITIVE_CALLER. (default: None)
Add another channel to represent HP tags per read. (default: None)
Auxiliary fields of the BAM/CRAM records are parsed. If either --sort-by-haplotypes or --add-hp-channel is set, then this option must also be set. (default: None)
If passed, the WES model file will be used. Only used in shortread mode. (default: None)
If True, include MED (default: None)
If True, allele counter left align INDELs for each read. (default: None)
Pileup image width. Only change this if you know your model supports this width. (default: 221)
If True, add insert_size channel into pileup image. By default, this parameter is true in WGS and WES mode. (default: None)
If True, don't add insert_size channel into the pileup image. (default: None)
Allow deepvariant to run on reads of size 512bp. The default size is 320 bp. (default: None)
Use an extra thread for the pre-align step. This parameter is more useful when --max-reads-size-512 is set. (default: None)
If True, allele counter keeps track of reads supporting ref. By default, allele counter keeps a simple count of the number of reads supporting ref. (default: None)
Calculate phases and add HP tag to all reads automatically. (default: None)
Minimum base quality in a k-mer sequence to consider. (default: 15)
Minimum distance between candidate windows for local assembly (default: 80)
If True, add gc (default: None)
If True, add hmer deletion quality channel into pileup image (default: None)
If True, add hmer insertion quality channel into pileup image (default: None)
If True, add non-hmer insertion quality channel into pileup image (default: None)
If True, ignore base quality channel. (default: None)
Comma-delimited list of auxiliary BAM fields to keep. Values can be [HP, tp, t0] (default: HP)
Hmer Indel alleles occurring at least this be advanced as candidates. Use this threshold if hmer and non-hmer indels should be treated differently (Ultima reads)Default will use the same threshold for hmer and non-hmer indels, as defined in vsc_min_fraction_indels. (default: None)
Add read-support from soft-clipped reads and other non-hmer insertion alleles,to the most frequent non-hmer insertion allele. (default: None)
If True, expect SB field in calls and write it to vcf (default: None)
Basecalling error for reference confidence model. (default: 0.001)
If true, add another channel to represent size of insertions. (good for flow-based sequencing) (default: None)
Max insertion size for ins_size_channel, larger insertions will look like max (have max intensity) (default: 10)
If using vcf_candidate_importer and multi-allelic sites are split across multiple lines in VCF, set to True so that variants are not grouped when transforming CallVariantsOutput to Variants. (default: None)
Ignore all input bam reads with size > 512bp (default: None)
Performance Options:
- --fq2bamfast
- --gpuwrite
- --gpuwrite-deflate-algo GPUWRITE_DEFLATE_ALGO
- --gpusort
- --use-gds
- --memory-limit MEMORY_LIMIT
- --low-memory
- --num-cpu-threads-per-stage NUM_CPU_THREADS_PER_STAGE
- --bwa-nstreams BWA_NSTREAMS
- --bwa-cpu-thread-pool BWA_CPU_THREAD_POOL
- --num-cpu-threads-per-stream NUM_CPU_THREADS_PER_STREAM
- --num-streams-per-gpu NUM_STREAMS_PER_GPU
- --run-partition
- --gpu-num-per-partition GPU_NUM_PER_PARTITION
- --max-reads-per-partition MAX_READS_PER_PARTITION
- --partition-size PARTITION_SIZE
- --read-from-tmp-dir
Use fq2bamfast as the alignment tool instead of fq2bam (default: None)
Use one GPU to accelerate writing final BAM. (default: None)
Choose the nvCOMP DEFLATE algorithm to use with --gpuwrite. Note these options do not correspond to CPU DEFLATE options. Valid options are 0 and 3. Option 0 is faster while option 3 provides a better compression ratio. (default=0) (default: None)
Use GPUs to accelerate sorting and marking. (default: None)
Use GPUDirect Storage (GDS) to enable a direct data path for direct memory access (DMA) transfers between GPU memory and storage. Must be used concurrently with --gpuwrite. Please refer to Parabricks Documentation > Best Performance for information on how to set up and use GPUDirect Storage. (default: None)
System memory limit in GBs during sorting and postsorting. By default, the limit is half of the total system memory. (default: 62)
Use low memory mode (default: None)
Number of CPU threads to use per stage. (default: 8)
Number of streams per GPU to use; note: more streams increases device memory usage (Argument only applies to --fq2bamfast) (default: 4)
Number of threads to devote to CPU thread pool per GPU (Argument only applies to --fq2bamfast) (default: 16)
Number of cpu threads to use per stream. (default: 6)
Number of streams to use per GPU. (default: 2)
Divide the whole genome into multiple partitions and run multiple processes at the same time, each on one partition. (default: None)
Number of GPUs to use per partition. (default: None)
The maximum number of reads per partition that are considered before following processing such as sampling and realignment. (default: 1500)
The maximum number of basepairs allowed in a region before splitting it into multiple smaller subregions. (default: 1000)
Running variant caller reading from bin files generated by Aligner and sort. Run postsort in parallel. This option will increase device memory usage. (default: None)
Common options:
- --logfile LOGFILE
- --tmp-dir TMP_DIR
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --no-seccomp-override
- --version
Path to the log file. If not specified, messages will only be written to the standard error output. (default: None)
Full path to the directory where temporary files will be stored.
Full path to the PetaGene installation directory. By default, this should have been installed at /opt/petagene. Use of this option also requires that the PetaLink library has been preloaded by setting the LD_PRELOAD environment variable. Optionally set the PETASUITE_REFPATH and PGCLOUD_CREDPATH environment variables that are used for data and credentials (default: None)
Do not delete the directory storing temporary files after completion.
Do not override seccomp options for docker (default: None).
View compatible software versions.
GPU options:
- --num-gpus NUM_GPUS
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used.
The --in-fq option takes the names of two FASTQ files, optionally followed by a quoted read group. The FASTQ filenames must not start with a hyphen.