Finetuning LLM in BioNeMo for a Downstream Task
Contents
Finetuning LLM in BioNeMo for a Downstream Task#
This notebook covers how to fine-tune MegaMolBART (MMB) for downstream task of predicting physicochemical properties of drugs.
One of the improtant tasks for chemoinformaticians is to develop models for predicting properties of small molecules.
These properties may include physicochemical parameters, such as lipophilicity, solubility, hydration free energy (LogP, LogD, and so on). It can also include certain pharmacokinetic/dynamic behaviors, such as Blood-Brain-Barrier/CNS permeability and Vd.
Modeling such properties can often become challenging along with choosing the appropriate and relevant descriptors/features for developing such prediction models.
In this notebook, we will use the encoder of pretrained Megamolbart model and add a MLP prediction head trained for physico-chemical parameter predictions.
Setup and Assumptions#
This tutorial assumes that the user has access to BioNeMo framework and NVIDIA’s BCP and DGX-Cloud compute infrastructure. The user is also expected to have required background details about
the BioNeMo framework, as described in the Quickstart Guide, and
running the model training jobs on BCP.
All model training and finetuning related commands should be executed inside the BioNeMo docker container.
The working directory needs to be /workspace/bionemo/examples/molecule/megamolbart for updating and running the following code.
In the section below, we will be using the one of the following datasets curated by MoleculeNet – ESOL dataset (https://moleculenet.org/datasets-1)
Lipophilicity: Experimental results of octanol/water distribution coefficient(logD at pH 7.4) [n=4200]
FreeSolv: Experimental and calculated hydration free energy of small molecules in water [n=642]
ESOL: Water solubility data(log solubility in mols per litre) for common organic small molecules [n=1129]
Example: Train Model for Compound Water Solubility (ESOL) Prediction using MMB Pretrained Model#
Launch BioNeMo development container
bash launch.sh dev
Locate physchem downstream task config in /workspace/bionemo/examples/molecule/megamolbart/conf/finetune_config.yaml
Download and preprocess dataset easily using BioNeMo yaml
#Set download location for datasets inside finetune_config.yaml
data:
dataset_path: /data/physchem
#Let's also ensure that our dataset is split to create training, validation and test sets
split_data: True
val_frac: 0.15 # proportion of samples used for validation set
test_frac: 0.15 # proportion of samples used for test set
Let’s ensure that we don’t try to train a model yet and instead run preprocess steps.
Simply set do_training to False inside our yaml config.
do_training: False
Now, lets run our downstream script and pass in the finetune_config.yaml
#assuming pwd: /workspace/bionemo/examples/molecule/megamolbart
python downstream_physchem.py --config-path=./conf/ --config-name=finetune_config
We should now have datasets for all three properties downloaded from MoleculeNet in our /data/phsychem folder.
Now’s we’ll specify which datasets we want to use to train our MLP prediction head by once again using our yaml config. This is done by simply setting model.data.task_name parameter to the name of the folder where we stored our ESOL dataset which is ‘delaney-processed’.
data:
task_name: SAMPL #specifies which MoleculeNet physchem dataset to use for training, expected values: SAMPL, Lipophilicity, or delaney-processed
Now, let’s indicated which column contains our SMILES string and our target value of interest for training the model based on the column headers in the csv file
data:
sequence_column: 'smiles'
target_column: 'measured log solubility in mols per litre'
#These parameters are all nested under the data key in the yaml file
Finally, we need to ensure that our script skips preprocessing and trains a model by setting do_training to True and we ensure that do_testing is set to True to also use of test dataset for evaluation.
do_training: True
do_testing: True
While we have already set reasonable defaults for the other model paramters necessary for training, it is important to note the parameters of the config.
The path to the pretrained megamolbart model should be already set:
restore_from_path: /model/molecule/megamolbart/megamolbart.nemo
Under the model parameters, we can set whether we want the encoder to be frozen or not, our micro batch size and other downstream task parameters used by the MLP prediction head
model:
encoder_frozen: True
micro_batch_size: 32
downstream_task:
n_outputs: 1
hidden_layer_size: 128
loss_func: MSELoss
Now, we are ready to train a model for our downstream task.
#assuming pwd: /workspace/bionemo/examples/molecule/megamolbart
python downstream_physchem.py --config-path=./conf/ --config-name=finetune_config
#A successful run should display summary statistics of training the model. An example of this is shown below:
wandb: Run summary:
wandb: consumed_samples 76800.0
wandb: epoch 100
wandb: global_step 2399.0
wandb: lr 0.00077
wandb: reduced_train_loss 0.17997
wandb: test_loss 0.47823
wandb: test_step_timing 0.00071
wandb: train_backward_timing 0.00076
wandb: train_step_timing 0.00416
wandb: trainer/global_step 2400
wandb: val_loss 0.42474
wandb: validation_step_timing 0.00153
Note
If the samples in the experiment folder are already processed, it will give an runtime error no samples left to consume. To avoid this error, users can either delete or move the experiment folder, or set ++exp_manager.resume_if_exists=false in the command line argument.
Now that we’ve trained on the ESOL dataset, to change which dataset we train is simple. To do a run where we train using the Lipophilicity dataset instead can be done as follows:
python downstream_physchem.py \
--config-path=./conf/ --config-name=finetune_config \
model.data.task_name=Lipophilicity \
model.data.target_column=exp \
++exp_manager.resume_if_exists=false
Results and Logging#
Results of your experiment, including model checkpoints, can then be found in /result/nemo_experiments/.
All logs from the experiment as well as the config parameters used to run that experiments are stores here as well.
[Optional] Setting up Weights and Biases account for tracking the model training process
If you would like to monitor the MegaMolBART model training process, set up Weights and Biases access by following the links:
For setting up the account: https://wandb.ai/site
Once the account is set, copy the API key: https://wandb.ai/authorize
Use this key in your .env file.
Monitoring the model training progress with Weights and Biases
Following are examples plots showing the model training run, as logged and plotted by weights and Biases (www.wandb.ai).
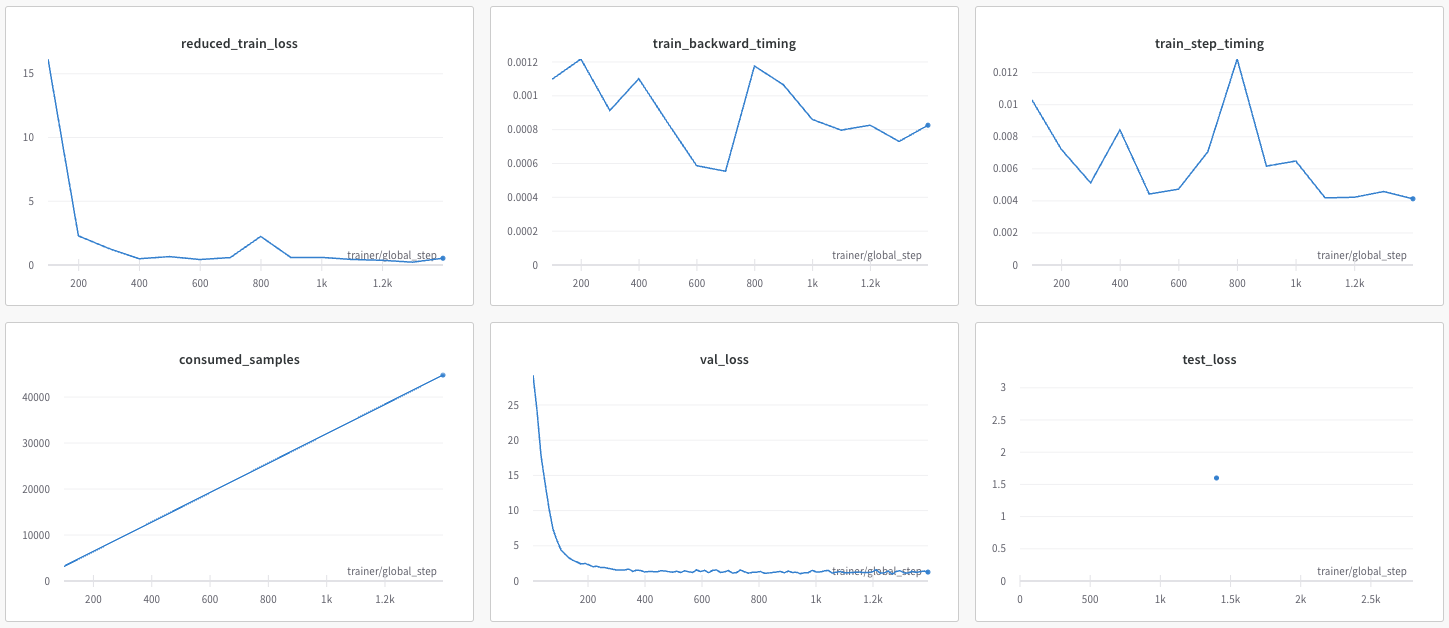
[Optional] Model parameters can also be changed by passing them as arguments to the script. This removes the need to edit and save the yaml config each time.
For example, we could run both the preprocessing step and model training using the commands below
#Data preprocessing
python downstream_physchem.py --config-path=./conf/ --config-name=finetune_config \
do_training=False \
model.data.dataset_path=/data/physchem \
model.data.split_data=True model.data.val_frac=0.15 \
model.data.test_frac=0.15
#Model Training for downstream task
python downstream_physchem.py --config-path=./conf/ --config-name=finetune_config \
do_training=True \
restore_from_path=/model/molecule/megamolbart/megamolbart.nemo \
model.encoder_frozen=True \
model.micro_batch_size=32 \
model.data.train_ds.data_file=/data/physchem/delaney-processed_splits/train.csv \
model.data.validation_ds.data_file=/data/physchem/delaney-processed_splits/val.csv \
model.data.test_ds.data_file=/data/physchem/delaney-processed_splits/test.csv