Overview#
DiffDock is a state-of-the-art generative model used for drug discovery that predicts the three-dimensional structure of a protein-ligand complex, a crucial step in the drug discovery process. It predicts the binding structure of a small molecule ligand to a protein, known as molecular docking or pose prediction.
DiffDock can:
Help AI drug discovery pipelines and open new research avenues for downstream task integrations.
Be highly accurate and computationally efficient.
Provide fast inference times and confidence estimates with high selective accuracy.
Training Dataset#
The DiffDock NIM model is trained on an expanded dataset that combines multiple high-quality sources:
PLINDER: A comprehensive protein-ligand interaction dataset curated from the Protein Data Bank (PDB), providing diverse real-world protein-ligand complexes for training.
SAIR (Structurally-Augmented IC50 Repository): The largest public dataset of protein–ligand 3D structures paired with binding potency measurements. SAIR, produced by SandboxAQ, contains over one million protein–ligand complexes (1,048,857 unique pairs) and a total of 5.2 million 3D structures, curated from the ChEMBL and BindingDB databases and cofolded using the Boltz-1x model. By providing this unprecedented scale of structure–activity data, SAIR enables researchers to train and evaluate new AI models for drug discovery by bridging the historical gap between molecular structure and drug potency prediction.
The combination of real experimental data from PLINDER and the large-scale structure–activity data from SAIR provides a more comprehensive training set, resulting in enhanced docking accuracy and better performance across diverse molecular systems.
Note
A more detailed description of the model can be found in the Model Card.
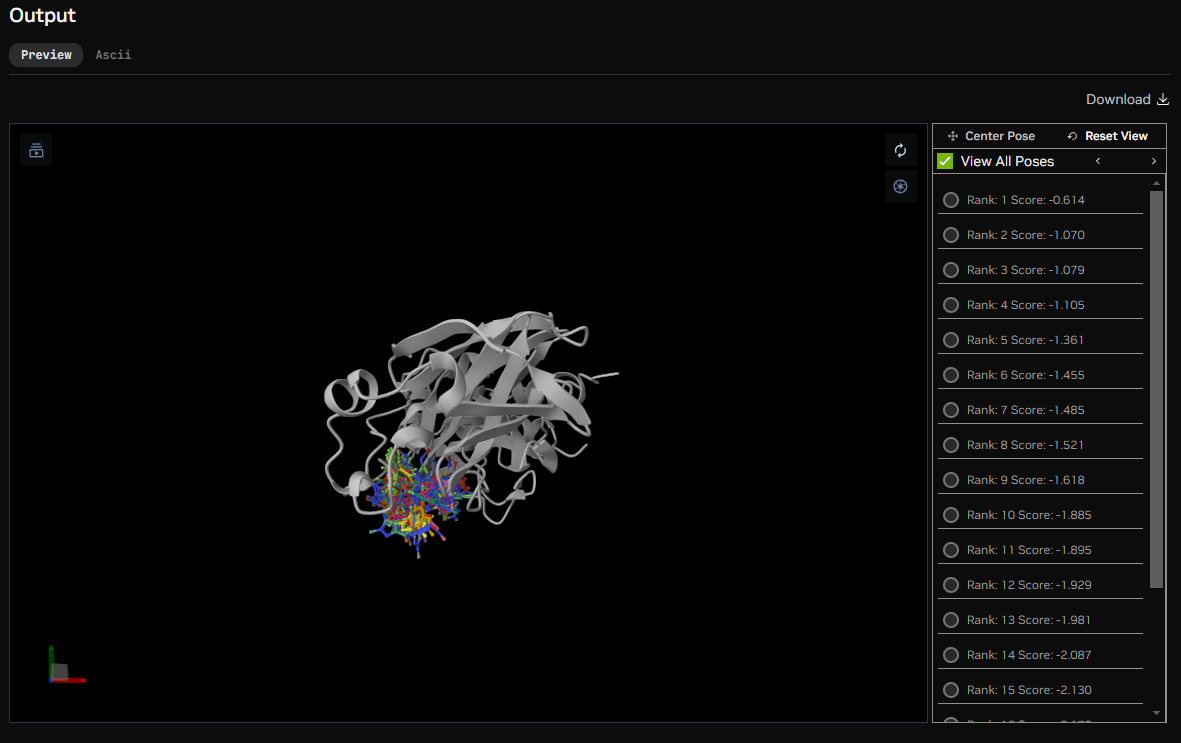
DiffDock is an equivariant geometric model for blind molecular docking pose estimation. It requires protein and molecule 3D structures as input and does not require any information about a binding pocket. During its diffusion process, the molecule’s position relative to the protein, its orientation, and the torsion angles can change. By running the learned reverse diffusion process, it transforms a distribution of noisy prior molecule poses to the one learned by the model. As a result, it outputs many sampled poses and ranks them via its confidence model.