denovomutation
Run the de novo mutation pipeline with three samples for de novo variant detection.
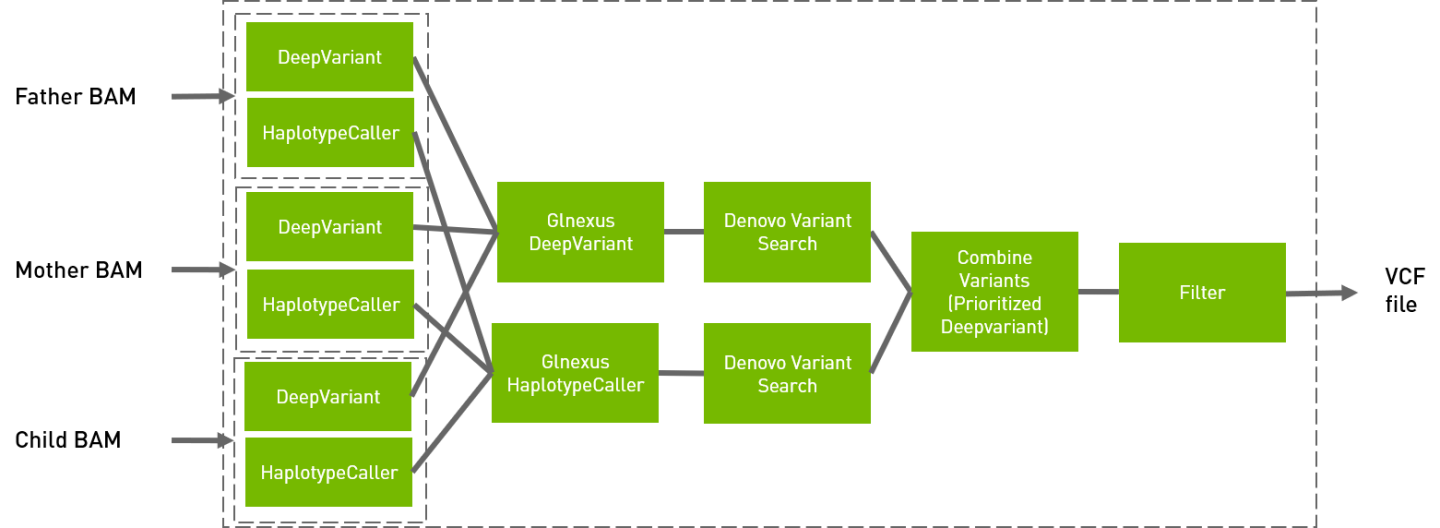
The following command runs the de novo pipeline.
$ pbrun denovomutation \
--ref Ref/Homo_sapiens_assembly38.fasta \
--in-mother-bam mother.bam \
--in-father-bam father.bam \
--in-child-bam child.bam \
--out-prefix output
(BETA) Run the de novo mutation pipeline with 3 samples for de novo variant detection.
Input/Output file options
- --ref REF
- --in-father-bam IN_FATHER_BAM
- --in-mother-bam IN_MOTHER_BAM
- --in-child-bam IN_CHILD_BAM
- --pb-model-file PB_MODEL_FILE
- --out-prefix OUT_PREFIX
Path to the reference file. (default: None)
Option is required.
Path to the input BAM/CRAM file of father for variant calling. This input file will be run through haplotypecaller and deepvariant. (default: None)
Option is required.
Path to the input BAM/CRAM file of mother for variant calling. This input file will be run through haplotypecaller and deepvariant. (default: None)
Option is required.
Path to the input BAM/CRAM file of child for variant calling. This input file will be run through haplotypecaller and deepvariant. (default: None)
Option is required.
Path of non-default parabricks model file for deepvariant. (default: None)
Prefix filename for output data. (default: None)
Option is required.
Options specific to this tool
- --disable-deepvariant
- --disable-haplotypecaller
- --disable-use-window-selector-model
- --haplotypecaller-options HAPLOTYPECALLER_OPTIONS
- --static-quantized-quals [STATIC_QUANTIZED_QUALS [STATIC_QUANTIZED_QUALS ...]]
- --disable-read-filter DISABLE_READ_FILTER
- --max-alternate-alleles MAX_ALTERNATE_ALLELES
- -G ANNOTATION_GROUP, --annotation-group ANNOTATION_GROUP
- -GQB [GVCF_GQ_BANDS [GVCF_GQ_BANDS ...]], --gvcf-gq-bands [GVCF_GQ_BANDS [GVCF_GQ_BANDS ...]]
- --rna
- --dont-use-soft-clipped-bases
- --ploidy PLOIDY
- --glnexus-options GLNEXUS_OPTIONS
Generate results for haplotypecaller only. (default: None)
Generate results for deepvariant only. (default: None)
Change the window selector model from Allele Count Linear to Variant Reads. This option will increase the accuracy and runtime. (default: None)
Pass supported haplotype caller options as one string. Currently supported original haplotypecaller options: -min-pruning <int>, -standard-min-confidence-threshold-for-calling <int>, -max-reads-per-alignment-start <int>, -min-dangling-branch-length <int>, -pcr-indel-model <NONE, HOSTILE, AGGRESSIVE, CONSERVATIVE>. e.g. --haplotypecaller-options="-min-pruning 4 -standard-min-confidence-threshold-for-calling 30". (default: -standard-min-confidence-threshold-for-calling 30)
Use static quantized quality scores to a given number of levels. Repeat this option multiple times for multiple bins. (default: None)
Disable the read filters for bam entries. Currently supported read filters that can be disabled: MappingQualityAvailableReadFilter, MappingQualityReadFilter, NotSecondaryAlignmentReadFilter, WellformedReadFilter. (default: None)
Maximum number of alternate alleles to genotype. (default: None)
Which groups of annotations to add to the output variant calls. Currently supported annotation groups: StandardAnnotation, StandardHCAnnotation, AS_StandardAnnotation. (default: None)
Exclusive upper bounds for reference confidence GQ bands. Must be in the range [1, 100] and specified in increasing order. (default: None)
Run haplotypecaller optimized for RNA Data. (default: None)
Don't use soft clipped bases for variant calling. (default: None)
Ploidy assumed for the bam file. Currently only haploid (ploidy 1) and diploid (ploidy 2) are supported. (default: 2)
Pass supported GLnexus options as one string. e.g. --glnexus-options="--list --squeeze". (default: None)
Common options:
- --logfile LOGFILE
- --tmp-dir TMP_DIR
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --no-seccomp-override
- --version
Path to the log file. If not specified, messages will only be written to the standard error output. (default: None)
Full path to the directory where temporary files will be stored.
Full path to the PetaGene installation directory. By default, this should have been installed at /opt/petagene. Use of this option also requires that the PetaLink library has been preloaded by setting the LD_PRELOAD environment variable. Optionally set the PETASUITE_REFPATH and PGCLOUD_CREDPATH environment variables that are used for data and credentials (default: None)
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in the installation directory.
Do not override seccomp options for docker (default: None).
View compatible software versions.
GPU options:
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used.
GPU devices to use for a run. By default, all GPU devices will be used.
To use specific GPU devices, enter a comma-separated list of GPU device
numbers. Possible device numbers can be found by examining the output of
the nvidia-smi command. For example, using --gpu-devices 0,1
would only use the first two GPUs.