umi_fgbio
Groups reads together that appear to have come from the same original molecule, based on the UMI of the read.
$ pbrun umi_fgbio \
--in-fq firstInput.fastq.gz secondInput.fastq.gz \
--ref theReferenceFile.fasta \
--out-dir directoryWithOutput \
--umi-in-header \
--strategy paired
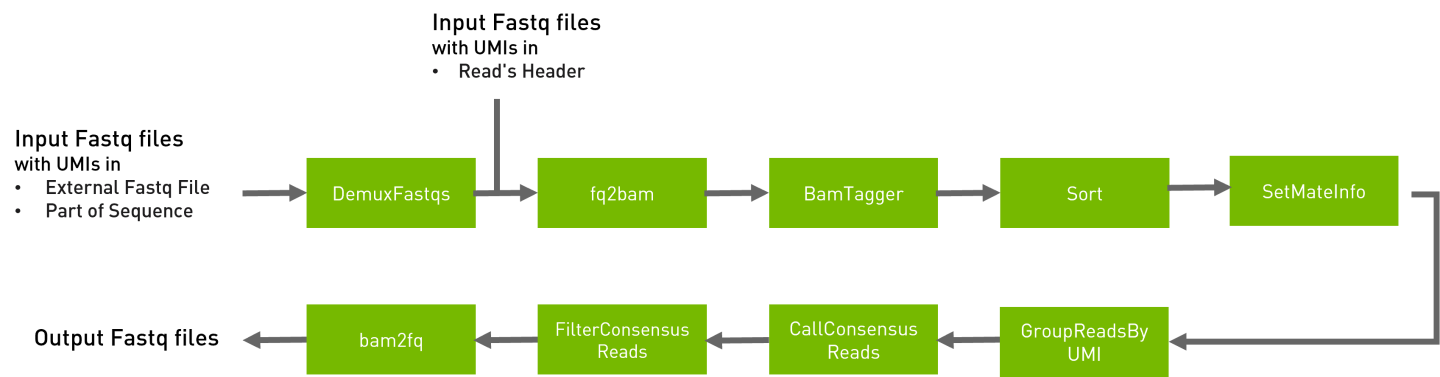
This UMI pipeline is based on Fulcrum Genomics toolkit, processes sequencing reads with molecular barcodes (also known as Unique Molecular Indices, UMIs),which provide impressive error correction and increased accuracy using a sequencing consensus read level.
Input/Output file options
- --in-fq [IN_FQ [IN_FQ ...]]
- --ref REF
- --metadata METADATA
- --out-dir OUT_DIR
Path to one or more fastq files each corresponding to a sub-read. Files can be in fastq or fastq.gz format. (default: None)
Option is required.
Path to the reference file (default: None)
Option is required.
Path to a file containing the metadata about the samples. If no file is provided, output reads will be put into unmatched files only. (default: None)
Path to the directory that will contain all of the generated files (default: None)
Option is required.
Options specific to this tool
- --umi-in-header
- -L INTERVAL, --interval INTERVAL
- --bwa-options BWA_OPTIONS
- --no-warnings
- --read-group-sm READ_GROUP_SM
- --read-group-lb READ_GROUP_LB
- --read-group-pl READ_GROUP_PL
- --read-group-id-prefix READ_GROUP_ID_PREFIX
- -ip INTERVAL_PADDING, --interval-padding INTERVAL_PADDING
- --read-structures [READ_STRUCTURES [READ_STRUCTURES ...]]
- --out-metrics OUT_METRICS
- --no-barcode
- --num-zip-threads NUM_ZIP_THREADS
- --num-sort-threads NUM_SORT_THREADS
- --max-records-in-ram MAX_RECORDS_IN_RAM
- --strategy STRATEGY
- --num-worker-threads NUM_WORKER_THREADS
- --min-reads MIN_READS
- --out-suffixF OUT_SUFFIXF
- --out-suffixF2 OUT_SUFFIXF2
- --out-suffixO OUT_SUFFIXO
- --out-suffixO2 OUT_SUFFIXO2
- --out-suffixS OUT_SUFFIXS
- --rg-tag RG_TAG
- --remove-qc-failure
- --num-threads NUM_THREADS
UMIs are in read header. (default: False)
Interval within which to call bqsr from the input reads. All intervals will have a padding of 100 to get read records and overlapping intervals will be combined. Interval files should be passed using the --interval-file option. This option can be used multiple times. e.g. "-L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000" (default: None)
Pass supported bwa mem options as one string. Current original bwa mem supported options, -M, -Y, -T. e.g. --bwa-options="-M -Y" (default: None)
Suppress warning messages about system thread and memory usage (default: None)
SM tag for read groups in this run (default: None)
LB tag for read groups in this run (default: None)
PL tag for read groups in this run (default: None)
prefix for ID and PU tag for read groups in this run. This prefix will be used for all pair of fastq files in this run. The ID and PU tag will consist of this prefix and an identifier which will be unique for a pair of fastq files (default: None)
Amount of padding (in base pairs) to add to each interval you are including. (default: None)
The read structure for each of the FASTQs. There must be one read structure per input fastq file. (default: None)
The file to which per-barcode metrics are written in the output directory. If none given, a file named demux_barcode_metrics.txt will be written to the output directory. (default: None)
Remove the requirement that input read structures must contain sample barcodes. (default: None)
Number of CPUs to use for zipping bam files in a run (default 16 for coordinate sorts and 10 otherwise) (default: None)
Number of CPUs to use for sorting in a run (default 10 for coordinate sorts and 16 otherwise) (default: None)
Maximum number of records in RAM when using a queryname or template coordinate sort mode; lowering this number will decrease maximum memory usage. (default: 65000000)
The UMI assignment strategy: adjacency or paired. (default: )
Option is required.
Number of threads for worker (default 14) (default: 14)
The minimum number of reads to produce a consensus base (default 1) (default: 1)
Output suffix used for paired reads that are first in pair. Suffix must end with ".gz" (default: _1.fastq.gz)
Output suffix used for paired reads that are second in pair. Suffix must end with ".gz" (default: _2.fastq.gz)
Output suffix used for orphan/unmatched reads that are first in pair. Suffix must end with ".gz". If no suffix is provided, these reads will be ignored (default: None)
Output suffix used for orphan/unmatched reads that are second in pair. Suffix must end with ".gz". If no suffix is provided, these reads will be ignored (default: None)
Output suffix used for single-end/unpaired reads. Suffix must end with ".gz". If no suffix is provided, these reads will be ignored (default: None)
Split reads into different fastq files based on the read group tag. Must be either PU or ID (default: None)
Remove reads from the output that have abstract QC failure (default: None)
Number of threads to run (default: 8)
Common options:
- --logfile LOGFILE
- --tmp-dir TMP_DIR
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --no-seccomp-override
- --version
Path to the log file. If not specified, messages will only be written to the standard error output. (default: None)
Full path to the directory where temporary files will be stored.
Full path to the PetaGene installation directory. By default, this should have been installed at /opt/petagene. Use of this option also requires that the PetaLink library has been preloaded by setting the LD_PRELOAD environment variable. Optionally set the PETASUITE_REFPATH and PGCLOUD_CREDPATH environment variables that are used for data and credentials (default: None)
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in the installation directory.
Do not override seccomp options for docker (default: None).
View compatible software versions.
GPU options:
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used.
GPU devices to use for a run. By default, all GPU devices will be used.
To use specific GPU devices, enter a comma-separated list of GPU device
numbers. Possible device numbers can be found by examining the output of
the nvidia-smi command. For example, using --gpu-devices 0,1
would only use the first two GPUs.