About Parabricks
NVIDIA Parabricks is a scalable genomics software suite for secondary analysis that provides GPU-accelerated versions of trusted, open-source tools. Compatible with leading sequencing instruments, Parabricks can be used across diverse bioinformatics workflows to increase speed and reduce cost—while enhancing accuracy and ensuring transparency. As a result, researchers can uncover biological insights faster.
Key Features
- Increase speed: Experience over 100x faster analysis of whole-genome sequencing (WGS) compared to CPU-only solutions. Reduce germline analysis from ~16 hours to under 10 minutes on 4 NVIDIA RTX PRO 6000 Server Edition GPUs.
- Reduce cost: Lower compute cost by up to 50% for WGS compared to CPU-only solutions.
- Enhance accuracy: Deploy high-accuracy deep learning for all major sequencers. Using accelerated deep learning tools like Google’s DeepVariant and DeepSomatic, Parabricks improves accuracy for germline and somatic variants.
- Ensure transparency: Reproduce results from trusted, open-source tools. Parabricks is publicly available and replicates open-source tools.
Learn more at the Parabricks developer page.
Performance
Time in minutes. These numbers were gathered by the NVIDIA Perflab team using Parabricks v4.7.0 with internal nodes. Only use them for reference. Speeds may vary depending on the data set, GPU instance, host CPU, memory availability, and other factors.
- 30× whole genome sequenced for Giraffe and DeepVariant with Illumina data.
- 35× whole genome sequenced for Minimap2 with Pacbio data.
- FQ2BAM_Meth (BWA-Meth) benchmarks downloaded whole-genome bisulfite sequencing data from the ENCODE portal (Sloan et al. 2016) with the following identifiers: ENCSR890UQO (library ENCLB898WPW).
- RNA_fq2bam (STAR) benchmarks used a Human Melanoma FASTQ sequenced on an Illumina NovaSeq 6000 (10x Genomics dataset).
How Can I Get Parabricks?
Parabricks is freely available as a public container on NGC for use on-premises or any cloud service platforms and providers. You can learn more about Parabricks on our webpage, including how to purchase enterprise support for Parabricks through NVIDIA AI Enterprise with guaranteed response times, priority security notifications and access to AI experts from NVIDIA.
Refer to the following Cloud Startup guides for more information on using Parabricks in the cloud:
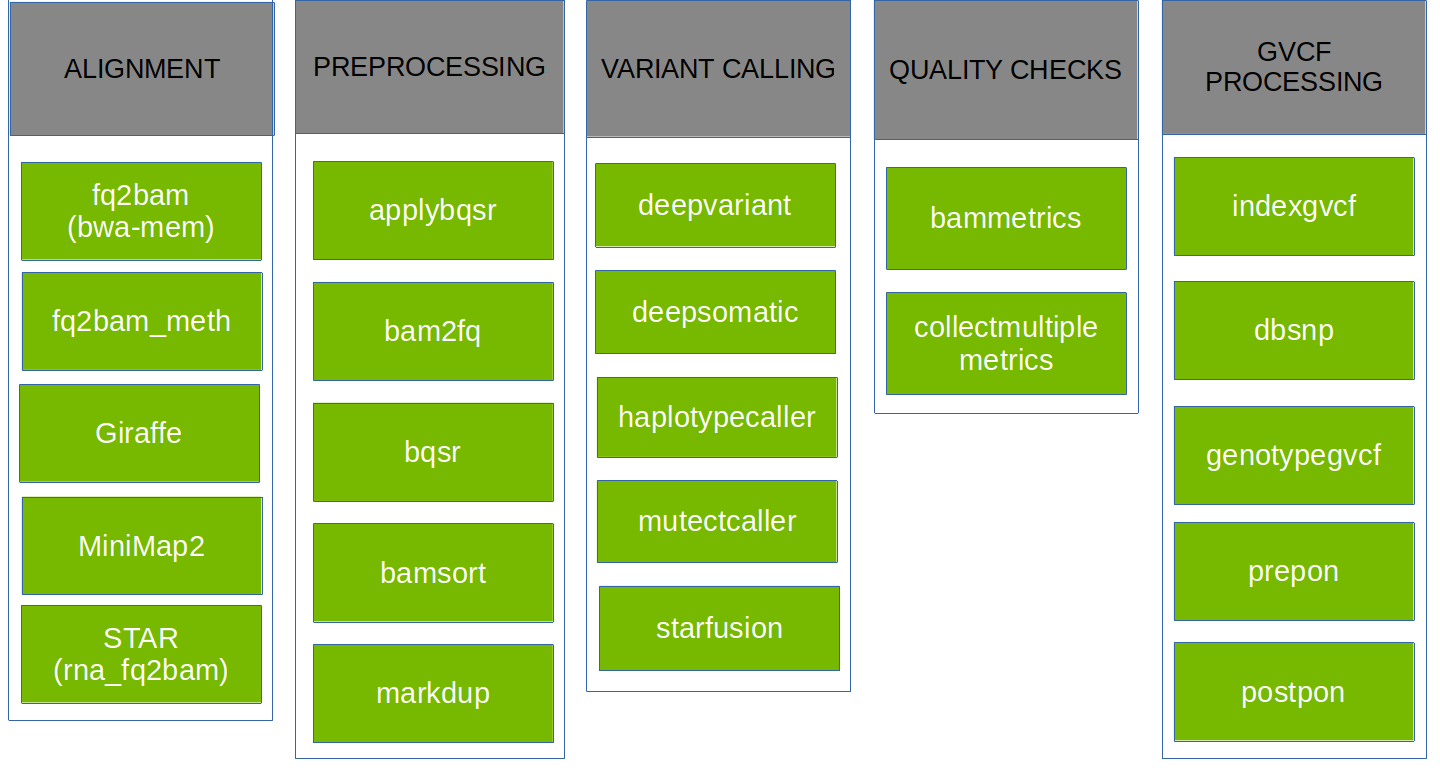
Available Tools
Parabricks supports the tools shown below:
Additionally, the Parabricks tool suite provides a number of variant-calling pipelines that are each a combination of several individual tools, combining into one tool what would otherwise be a multi-step process. These pipelines are shown below: