NVIDIA CLARA PARABRICKS DOCUMENTATION
Parabricks is a software suite for performing secondary analysis of next generation sequencing (NGS) DNA and RNA data. A major benefit of Parabricks is that it is designed to deliver results at blazing fast speeds and low cost. Parabricks can analyze whole human genomes in about 45 minutes, compared to about 30 hours for 30x WGS data. The best part is the output results exactly match the commonly used software. So, it’s fairly simple to verify the accuracy of the output.
Under the hood, it achieves this performance through tight integration with GPUs, which excel at performing data parallel computation much more effectively than traditional CPU-based solutions. Parabricks was built from the ground up by GPU computing and Deep Learning experts who wanted to develop the fastest and most efficient possible implementation of common genomics algorithms used in secondary analysis.
You can learn more at https://developer.nvidia.com/clara-parabricks
Parabricks is a software suite for genomic analysis. It delivers major improvements in throughput time for common analytical tasks in genomics, including germline and somatic analysis. The core of the Parabricks software is its data pipeline which takes raw data and transforms it according to the user’s requirements.
The Parabricks software supports the Tools shown below:
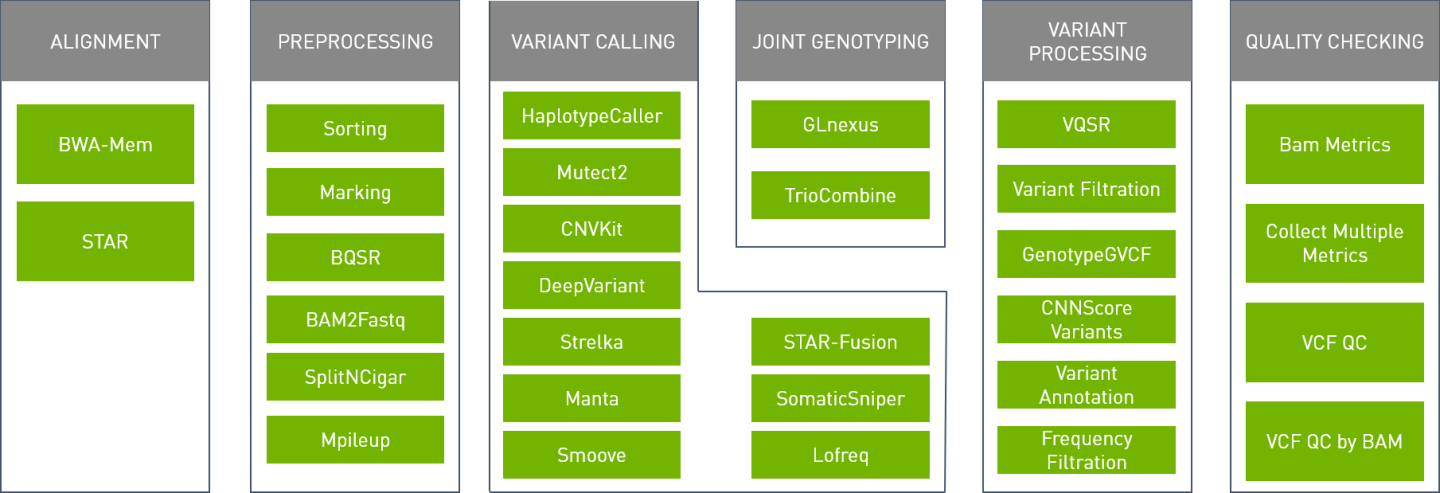
It supports the Pipelines shown below:

The Parabricks software can be configured to run specific accelerated tools or run full pipelines that are commonly used. The standalone tools page covers individual tools and the pipelines page discuses how to run commonly used pipelines.
NVIDIA Parabricks’ pipelines have been tested on Dell, HPE, IBM, and NVIDIA servers at Amazon Web Services, Google Cloud, Oracle Cloud Infrastructure, and Microsoft Azure.
The following standalone tools can be used with the NVIDIA Clara Parabricks Pipelines software. Please click on the tool names for tool specific options.
Tool |
Details |
Align using bwa-mem, co-ordinate sort and mark duplicates, optionally you can run bqsr. |
|
Generate a BQSR report on a bam file. |
|
Apply a BQSR report on a bam file to generate new bam file. |
|
GPU-HaplotypeCaller for calling germline variants. |
|
Accelerated samtools mpileup to generate pileup from a bam file. |
|
Accelerated bcftools mpileup to generate pileup from a bam file. |
|
Accelerated bcftools call variant caller. |
|
Accelerated Somaticsniper for tumor-normal analysis. |
|
Somaticsniper workflow to generate VCF from BAM input files. |
|
Structural variant (SV) and indel caller from mapped paired-end sequencing reads. |
|
SNP and indel caller from mapped paired-end sequencing reads. |
|
Strelka workflow to generate VCF from BAM/CRAM input files. |
|
GPU-Mutect2 for tumor-normal analysis. |
|
GPU-DeepVariant for calling germline variants. |
|
Accelerated copy number variant caller. |
|
Collect WGS Metrics on a bam file. |
|
Collect multiple classes of metrics for a bam file. |
|
Annotate variants based on a variant database. |
|
Filter variants using Convolutional Neural Network. |
|
Build a recalibration model to score variant quality and apply a score cutoff to filter variants. |
|
Combine gVCF of 2 or 3 samples. |
|
Scalable gVCF merging and joint variant calling for population sequencing projects. |
|
Index a VCF/gVCF file. |
|
Convert a gVCF To VCF. |
|
Mapping RNA reads to a reference, using a two-pass mode to get better alignments around novel splice junctions. |
|
Uses the STAR aligner to identify candidate fusion transcripts. |
Here are the versions that you can find in this package
Tool |
Version |
bwa |
0.7.15 |
GATK |
4.1.0.0 |
STAR |
2.7.2a |
STAR-Fusion |
1.7.0 |
Deepvariant |
1.0.0 |
lofreq |
2.1.5 |
somaticsniper |
1.0.5.0 |
cnvkit |
0.9.7 |
glnexus |
1.2.7 |
strelka |
2.9.0 |
manta |
1.6.0 |
samtools |
1.10 |
bcftools |
1.10.2 |
The following standalone pipelines can be used with the NVIDIA Clara Parabricks Pipelines software. Please click on the tool names for tool specific options.
Pipeline |
Functionality |
Detect germline variants for a given sample from fastq files. Compatible with GATK4 best practices. |
|
Detect germline variants for a given sample from fastq files using DeepVariant caller. |
|
Detect germline variants for a given sample from fastq files with correct ploidy values for human sex chromosome handling. Compatible with GATK4 best practices. |
|
Detect somatic variants for a given tumor/normal fastq files. Compatible with GATK4 best practices. |
|
Detect RNAseq short variants (SNPs + Indels). Compatible with GATK4 best practices. |
|
(BETA) De novo mutation pipeline with 3 samples for de novo variant detection |