rna_gatk
Run GATK best practices for RNAseq short variant discovery (SNPs + Indels).
The RNA GATK pipeline processes the input FASTQ files, performing the steps show below. The output is in VCF format.
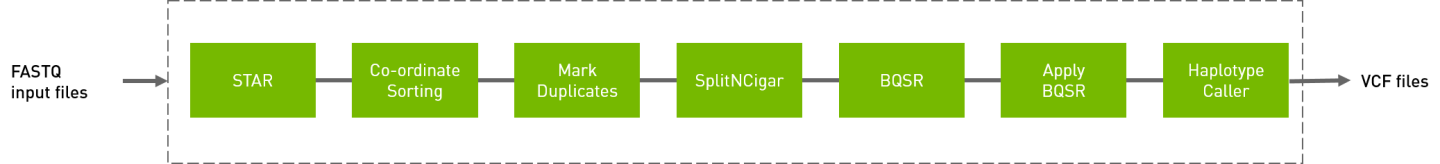
# The command line below will run the RNA GATK pipeline.
$ pbrun rna_gatk \
--ref Ref/Homo_sapiens_assembly38.fasta \
--in-fq Data/sample_1.fq.gz Data/sample_2.fq.gz \
--read-files-command zcat \
--genome-lib-dir Ref/ \
--out-variants output.vcf \
--out-bam tumor.bam \
--output-dir output
Run the GATK Best Practices pipeline for RNA-seq data from FASTQ to VCF.
Input/Output file options
- --ref REF
- --in-fq [IN_FQ [IN_FQ ...]]
- --in-se-fq [IN_SE_FQ [IN_SE_FQ ...]]
- --genome-lib-dir GENOME_LIB_DIR
- --knownSites KNOWNSITES
- --interval-file INTERVAL_FILE
- --output-dir OUTPUT_DIR
- --out-recal-file OUT_RECAL_FILE
- --out-bam OUT_BAM
- --out-variants OUT_VARIANTS
Path to the reference file. (default: None)
Option is required.
Path to the pair-ended FASTQ files followed by optional read groups with quotes (Example: "@RGtID:footLB:lib1tPL:bartSM:sampletPU:foo"). The files must be in fastq or fastq.gz format. All sets of inputs should have a read group; otherwise, none should have a read group, and it will be automatically added by the pipeline. This option can be repeated multiple times. Example 1: --in-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz --in-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz. Example 2: --in-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz "@RGtID:footLB:lib1tPL:bartSM:sampletPU:unit1" --in-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz "@RGtID:foo2tLB:lib1tPL:bartSM:sampletPU:unit2". For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU. (default: None)
Path to the single-ended FASTQ file followed by optional read group with quotes (Example: "@RGtID:footLB:lib1tPL:bartSM:sampletPU:foo"). The file must be in fastq or fastq.gz format. Either all sets of inputs have read group or none should have it and will be automatically added by the pipeline. This option can be repeated multiple times. Example 1: --in-se-fq sampleX_1.fastq.gz --in-se-fq sampleX_2.fastq.gz . Example 2: --in-se-fq sampleX_1.fastq.gz "@RGtID:footLB:lib1tPL:bartSM:sampletPU:unit1" --in-se-fq sampleX_2.fastq.gz "@RGtID:foo2tLB:lib1tPL:bartSM:sampletPU:unit2" . For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU (default: None)
Path to a genome resource library directory. The indexing required to run STAR should be completed by the user beforehand. (default: None)
Option is required.
Path to a known indels file. The file must be in vcf.gz format. This option can be used multiple times. (default: None)
Path to an interval file in one of these formats: Picard-style (.interval_list or .picard), GATK-style (.list or .intervals), or BED file (.bed). This option can be used multiple times. (default: None)
Path to the directory that will contain all of the generated files. (default: None)
Option is required.
Path of the report file after Base Quality Score Recalibration. (default: None)
Path of the output BAM file. (default: None)
Option is required.
Path of the vcf/g.vcf/gvcf.gz file after variant calling. The argument can also be a local folder in batch mode. (default: None)
Option is required.
Tool Options:
- --num-cpu-threads NUM_CPU_THREADS
- --no-ignore-mark
- --num-threads NUM_THREADS
- --out-prefix OUT_PREFIX
- --read-files-command READ_FILES_COMMAND
- --read-group-sm READ_GROUP_SM
- --read-group-lb READ_GROUP_LB
- --read-group-pl READ_GROUP_PL
- --read-group-id-prefix READ_GROUP_ID_PREFIX
- --two-pass-mode TWO_PASS_MODE
- --read-length READ_LENGTH
- --haplotypecaller-options HAPLOTYPECALLER_OPTIONS
- --static-quantized-quals STATIC_QUANTIZED_QUALS
- --gvcf
- --batch
- --disable-read-filter DISABLE_READ_FILTER
- --max-alternate-alleles MAX_ALTERNATE_ALLELES
- -G ANNOTATION_GROUP, --annotation-group ANNOTATION_GROUP
- -GQB GVCF_GQ_BANDS, --gvcf-gq-bands GVCF_GQ_BANDS
- --rna
- --dont-use-soft-clipped-bases
- --run-partition
- --no-alt-contigs
- --ploidy PLOIDY
- -L INTERVAL, --interval INTERVAL
- -ip INTERVAL_PADDING, --interval-padding INTERVAL_PADDING
Number of CPU threads to traverse separate chromosomes in splitncigar. (default: 6)
Do not ignore marked reads in sorted output. (default: None)
Number of running worker threads per GPU. (default: 4)
Prefix filename for output data. (default: None)
Command line to execute for each of the input files. This command should generate FASTA or FASTQ text and send it to stdout: For example: zcat to uncompress .gz files, bzcat to uncompress .bz2 files, etc. (default: None)
SM tag for read groups in this run. (default: None)
LB tag for read groups in this run. (default: None)
PL tag for read groups in this run. (default: None)
Prefix for the ID and PU tags for read groups in this run. This prefix will be used for all pairs of FASTQ files in this run. The ID and PU tags will consist of this prefix, and an identifier that will be unique for a pair of FASTQ files. (default: None)
Two-pass mapping mode. The string can be "None" for one-pass mapping or "Basic" for basic two-pass mapping with all 1st pass junctions inserted into the genome indices on the fly. (default: Basic)
Input read length used to determine sjdbOverhang. (default: None)
Pass supported haplotype caller options as one string. Currently supported original haplotypecaller options: -min-pruning <int>, -standard-min-confidence-threshold-for-calling <int>, -max-reads-per-alignment-start <int>, -min-dangling-branch-length <int>, -pcr-indel-model <NONE, HOSTILE, AGGRESSIVE, CONSERVATIVE>, --output-mode <EMIT_VARIANTS_ONLY, EMIT_ALL_CONFIDENT_SITES, EMIT_ALL_ACTIVE_SITES> (e.g. --haplotypecaller-options="-min-pruning 4 -standard-min-confidence-threshold-for-calling 30"). (default: None)
Use static quantized quality scores to a given number of levels. Repeat this option multiple times for multiple bins. (default: None)
Generate variant calls in .gvcf Format. (default: None)
Given an input list of BAMs, run the variant calling of each BAM using one GPU, and process BAMs in parallel based on how many GPUs the system has. (default: None)
Disable the read filters for BAM entries. Currently, the supported read filters that can be disabled are MappingQualityAvailableReadFilter, MappingQualityReadFilter, NotSecondaryAlignmentReadFilter, and WellformedReadFilter. (default: None)
Maximum number of alternate alleles to genotype. (default: None)
The groups of annotations to add to the output variant calls. Currently supported annotation groups are: StandardAnnotation, StandardHCAnnotation, and AS_StandardAnnotation. (default: None)
Exclusive upper bounds for reference confidence GQ bands. Must be in the range [1, 100] and specified in increasing order. (default: None)
Run haplotypecaller optimized for RNA data. (default: None)
Don't use soft clipped bases for variant calling. (default: None)
Divide the whole genome into multiple partition and run multiple processes at the same time, each on 1 partition. (default: None)
Get rid of output records for alternate contigs. (default: None)
Ploidy assumed for the BAM file. Currently only haploid (ploidy 1) and diploid (ploidy 2) are supported. (default: 2)
Interval within which to call the variants from the BAM/CRAM file. All intervals will have a padding of 100 to get read records, and overlapping intervals will be combined. Interval files should be passed using the --interval-file option. This option can be used multiple times (e.g. "-L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000"). (default: None)
Amount of padding (in base pairs) to add to each interval you are including. (default: None)
Common options:
- --logfile LOGFILE
- --tmp-dir TMP_DIR
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --no-seccomp-override
- --version
Path to the log file. If not specified, messages will only be written to the standard error output. (default: None)
Full path to the directory where temporary files will be stored.
Full path to the PetaGene installation directory. By default, this should have been installed at /opt/petagene. Use of this option also requires that the PetaLink library has been preloaded by setting the LD_PRELOAD environment variable. Optionally set the PETASUITE_REFPATH and PGCLOUD_CREDPATH environment variables that are used for data and credentials (default: None)
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in the installation directory.
Do not override seccomp options for docker (default: None).
View compatible software versions.
GPU options:
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used.
GPU devices to use for a run. By default, all GPU devices will be used.
To use specific GPU devices, enter a comma-separated list of GPU device
numbers. Possible device numbers can be found by examining the output of
the nvidia-smi command. For example, using --gpu-devices 0,1
would only use the first two GPUs.
The --in-fq option takes the names of two FASTQ files, optionally followed by a quoted read group. The FASTQ filenames must not start with a hyphen.
In the values provided to --haplotypecaller-options --output-mode requires two leading hyphens, while all other values take a single hyphen.