Whole-Genome Somatic Small Variant Calling
This how-to explores calling variants using Mutect2 in the SEQC2 Consortium Sample.
Samtools and BWA
Parabricks
GATK4 v4.2.0.0
bedtools
R (vcfR, UpSetR)
At least 512GB of disk space (preferably on “balanced” or SSD storage)
At least two NVIDIA Tesla T4 GPUs; the commands and time estimates below utilize four NVIDIA Tesla T4 GPUs.
At least 24 CPU cores; the commands below use 48 vCPU cores.
At least 48GB of RAM per GPU
A functional Parabricks install (version 4.0 or newer)
The examples below use an AWS VM g4dn.12xlarge with 48 vCPU cores, 192GB of RAM, and 4 NVIDIA Tesla T4 GPUs running Ubuntu 18.04.5 LTS.
This how-to runs through a full Whole Genome Sequencing (WGS) somatic variant analysis pipeline for calling SNPs, MNPs, and small indels on real 30X short-read human data. Such analyses are commonly used in cancer genomics studies. For WGS somatic variant analysis, you will utilize the example data generated by "The Somatic Mutation Working Group of the SEQC2 Consortium". The dataset contains multiplatform sequencing from HCC1395, which is a triple negative breast cancer cell line, and HCC1395 BL, which is the matched normal cell line. The SEQC2 dataset contains whole genome (WGS) and Exome, sequencing performed at six different sequencing centers, and multiple replicates to minimized potential biases from sequencing technologies/assays. Furthermore, the dataset was processed through nine bioinformatics pipelines to evaluate accuracy and reproducibility. The SEQC2 consortium reports artifacts generated due to sample and library processing, along with evaluating the capabilities and limitations of bioinformatics tools for artifact detection and removal. A series of detailed articles are available below:
Establishing reference samples for detection of somatic mutations and germline variants with NGS technologies (accepted Nat. Biotechnol.)
Achieving reproducibility and accuracy in cancer mutation detection with whole-genome and whole-exome sequencing (accepted Nat. Biotechnol.)
Robust Cancer Mutation Detection with Deep Learning Models Derived from Tumor-Normal Sequencing Data (under revision)
Whole Genome and Exome Sequencing Reference Datasets from A Multi-center and Cross-platform Benchmark Study (under revision)
Personalized genome assembly for accurate cancer somatic mutation discovery using cancer-normal paired reference samples (under revision)
Comprehensive Assessment of Somatic Copy Number Variation Calling Using Next-Generation Sequencing Data (under revision)
The SEQC2 dataset is well characterized and the "Truth Variant Call set" provided by the consortium allows you to validate the results from the WGS somatic variant analysis pipeline. When you finish variant calling, you will annotate the VCF with several databases to determine which variants are common or might be associated with disease. You will filter out common variants (those observed frequently in 1000 Genomes) and then use NVIDIA Parabricks tools for quality control to assess the variant caller results.
The example dataset used for this how-to resides on NCBI SRA and can be
downloaded using wget. Once the SRA files are downloaded, install the SRA Toolkit
to convert SRA files to paired-end FASTQ files using the instructions
given below. Note that SRR7890827 is a normal sample and SRR7890824 is a tumor
sample from the HCC1395 cell line. Paired-end WGS sequencing was performed for
each sample on an Illumina HiSeq X.
## Download publicly available SRA files using wget. Both files are
# about 65 GB in size.
# Normal sample
wget https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR7890827/SRR7890827 --output-document=SRR7890827.sra
# Tumor sample
wget https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR7890824/SRR7890824 --output-document=SRR7890824.sra
## Convert SRA to FASTQ files
fastq-dump --split-files ./SRR7890827.sra --gzip
fastq-dump --split-files ./SRR7890824.sra --gzip
Once the SRA files have been converted to FASTQ format, they are no longer needed and may be deleted.
If you have your own data, you’ll likely be able to substitute it directly into the commands in this how-to to get accurate, multi-caller variant calls.
Next, download and index a reference genome to use in alignment and variant calling. You'll use GRCh38 for alignment, which may be downloaded from this page:
From that website, download and extract the contents of:
GRCh38.d1.vd1.fa.tar.gz
If you don't want to build your own BWA indices, also download and extract the contents of:
GRCh38.d1.vd1_BWA.tar.gz and
GRCh38.d1.vd1_GATK_indices.tar.gz
Otherwise you'll need to build your own indices. Instructions for building the indices are given below.
You’ll need these additional VCF files and their indices:
Additionally, you'll need BED and VCF files for the truth set:
If you use a reference file for which the index is not available, you can index it using BWA. Indexing should take 30 to 60 minutes.
tar -xvzf GRCh38.d1.vd1.fa.tar.gz
## Note this is baseline bwa.
bwa index GRCh38.d1.vd1.fa
The pbrun fq2bam command runs read alignment, as well as sorting, duplicate marking, and base quality score recalibration (BQSR) according to GATK best practices, but at a much faster rate than community tools by leveraging up to eight NVIDIA GPUs. The pbrun fq2bam command also generates a BQSR report, which is used to improve the base qualities within the BAM files and is used by MuTect2 downstream for variant calling. Note that this how-to adds a custom read group to make the sample easier to identify. If the sample is later merged with others in downstream analysis, adding a custom read group will be essential when running alignment for matched tumor/normal or multi-sample analyses. You will also need to pass a set of known sites to generate the BQSR report:
GCF_000001405.39.gz
ALL.wgs.1000G_phase3.GRCh38.ncbi_remapper.20150424.shapeit2_indels.vcf.gz
Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz
## Aligning Normal sample FASTQ file
$ docker run \
--gpus all \
--workdir /workdir \
--rm \
--volume $(pwd):/workdir \
--volume $(pwd):/outputdir \
pbrun fq2bam \
--ref /workdir/GRCh38.d1.vd1.fa \
--in-fq /workdir/SRR7890827_1.fastq.gz /workdir/SRR7890827_2.fastq.gz "@RG\tID:id_SRR7890827_rg1\tLB:lib1\tPL:bar\tSM:sm_SRR7890827\tPU:pu_SRR7890827_rg1" \
--knownSites /workdir/Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz \
--knownSites /workdir/GCF_000001405.39.gz \
--knownSites /workdir/ALL.wgs.1000G_phase3.GRCh38.ncbi_remapper.20150424.shapeit2_indels.vcf.gz \
--out-recal-file /outputdir/SRR7890827-WGS_FD_N_BQSR_REPORT.txt \
--bwa-options=-Y \
--out-bam /outputdir/SRR7890827-WGS_FD_N.bam
## Aligning Tumor sample FASTQ file
$ docker run \
--gpus all \
--workdir /workdir \
--rm \
--volume $(pwd):/workdir \
--volume $(pwd):/outputdir \
pbrun fq2bam \
--ref /workdir/GRCh38.d1.vd1.fa \
--in-fq /workdir/SRR7890824_1.fastq.gz /workdir/SRR7890824_2.fastq.gz "@RG\tID:id_SRR7890824_rg1\tLB:lib1\tPL:bar\tSM:sm_SRR7890824\tPU:pu_SRR7890824_rg1" \
--knownSites /workdir/Mills_and_1000G_gold_standard.indels.b38.primary_assembly.vcf.gz \
--knownSites /workdir/GCF_000001405.39.gz \
--knownSites /workdir/ALL.wgs.1000G_phase3.GRCh38.ncbi_remapper.20150424.shapeit2_indels.vcf.gz \
--out-recal-file /outputdir/SRR7890824-WGS_FD_T_BQSR_REPORT.txt \
--bwa-options=-Y \
--out-bam /outputdir/SRR7890824-WGS_FD_T.bam
The pbrun fq2bam command performs read alignment, sorting, duplicate marking and indexing; it should take about 50 minutes running on four NVIDIA Tesla T4 GPUs. The output of fq2bam is a BAM file, a BAI index, and a BQSR report file. These files will be used as inputs to the multiple SNP/indel callers run in the next step.
Parabricks currently supports five variant callers: Mutect2, MuSE, LoFreq, Somatic Sniper, and Strelka. Each uses slightly different methods for calling variants, with trade-offs in sensitivity and specificity for each. This example will run pbrun mutectcaller (Mutect2) to generate a set of small variant calls:
$ docker run \
--gpus all \
--workdir /workdir \
--rm \
--volume $(pwd):/workdir \
--volume $(pwd):/outputdir \
pbrun mutectcaller \
--ref /workdir/GRCh38.d1.vd1.fa \
--in-tumor-bam /workdir/SRR7890824-WGS_FD_T.bam \
--in-normal-bam /workdir/SRR7890827-WGS_FD_N.bam \
--in-tumor-recal-file S/workdir/RR7890824-WGS_FD_T_BQSR_REPORT.txt \
--in-normal-recal-file /workdir/SRR7890827-WGS_FD_N_BQSR_REPORT.txt \
--out-vcf /outputdir/SRR7890824-SRR7890827-WGS_FD.vcf \
--tumor-name sm_SRR7890824 \
--normal-name sm_SRR7890827
This command puts the called variants in SRR7890824-SRR7890827-WGS_FD.vcf and creates
the SRR7890824-SRR7890827-WGS_FD.vcf.stats file, which will be used in subsequent steps.
Looking for variants of clinical interest, you might consider running the other variant callers and taking the intersection of two or more callers to generate a highly specific callset. On the other hand, if you are less concerned with false positives but need a highly-sensitive callset, you could take the union of all five callers. Such vote-based consensus approaches have been employed in numerous large studies to address the shortcomings of individual callers.
First run MuTect2, which is the most widely used variant caller developed at Broad. Parabricks provides an accelerated version of MuTect2 v4.2.0.0 that is 10-15X faster than the community version. MuTect2 should run in under 30 minutes on a four-T4 GPU system.
The results of MuTect2 are plain-text VCF files.
Next, process the Mutect2 VCF files to extract non-indel variants using the GATK4 SelectVariants tool, which makes it possible to select a subset of variants based on various criteria in order to facilitate certain analyses. This example analysis extracts only the SNV and MNV variants, excluding small indels for further downstream validation of the variant calls in comparison to truthset (see detailed instructions below):
$ java -jar gatk-4.2.0.0/gatk-package-4.2.0.0-local.jar FilterMutectCalls \
-O SRR7890824-SRR7890827-WGS_FD.FilterMutectCalls.vcf \
-R GRCh38.d1.vd1.fa \
-V SRR7890824-SRR7890827-WGS_FD.vcf
Once you have the SNV/MNV .vcf file, use GATK4 FilterMutectCalls to apply filters
to the raw output of Mutect2. Parameters contained in M2FiltersArgumentCollection are
described here.
The following example uses a high confidence region bedfile to intersect the Mutect2 variants calls in the high confidence region and use the PASS filter variants for validation.
$ java -jar gatk-4.1.0.0/gatk-package-4.1.0.0-local.jar SelectVariants \
-R GRCh38.d1.vd1.fa \
-V SRR7890824-SRR7890827-WGS_FD.FilterMutectCalls.vcf \
--select-type-to-exclude INDEL \
-O SRR7890824-SRR7890827-WGS_FD.SNV-MNV.FilterMutectCalls.vcf
$ bedtools intersect \
-header \
-a SRR7890824-SRR7890827-WGS_FD.SNV-MNV.FilterMutectCalls.vcf \
-b High-Confidence_Regions_v1.2.bed > SRR7890824-SRR7890827-WGS_FD.SNV-MNV.FilterMutectCalls.hc.vcf
$ grep "#" SRR7890824-SRR7890827-WGS_FD.SNV-MNV.FilterMutectCalls.hc.vcf > mutect_header.txt
$ grep -v "#" SRR7890824-SRR7890827-WGS_FD.SNV-MNV.FilterMutectCalls.hc.vcf | awk '{if ($7 == "PASS")print}' > mutect_body.txt
$ cat mutect_header.txt mutect_body.txt > SRR7890824-SRR7890827-WGS_FD.SNV-MNV.FilterMutectCalls.hc.PASS.vcf
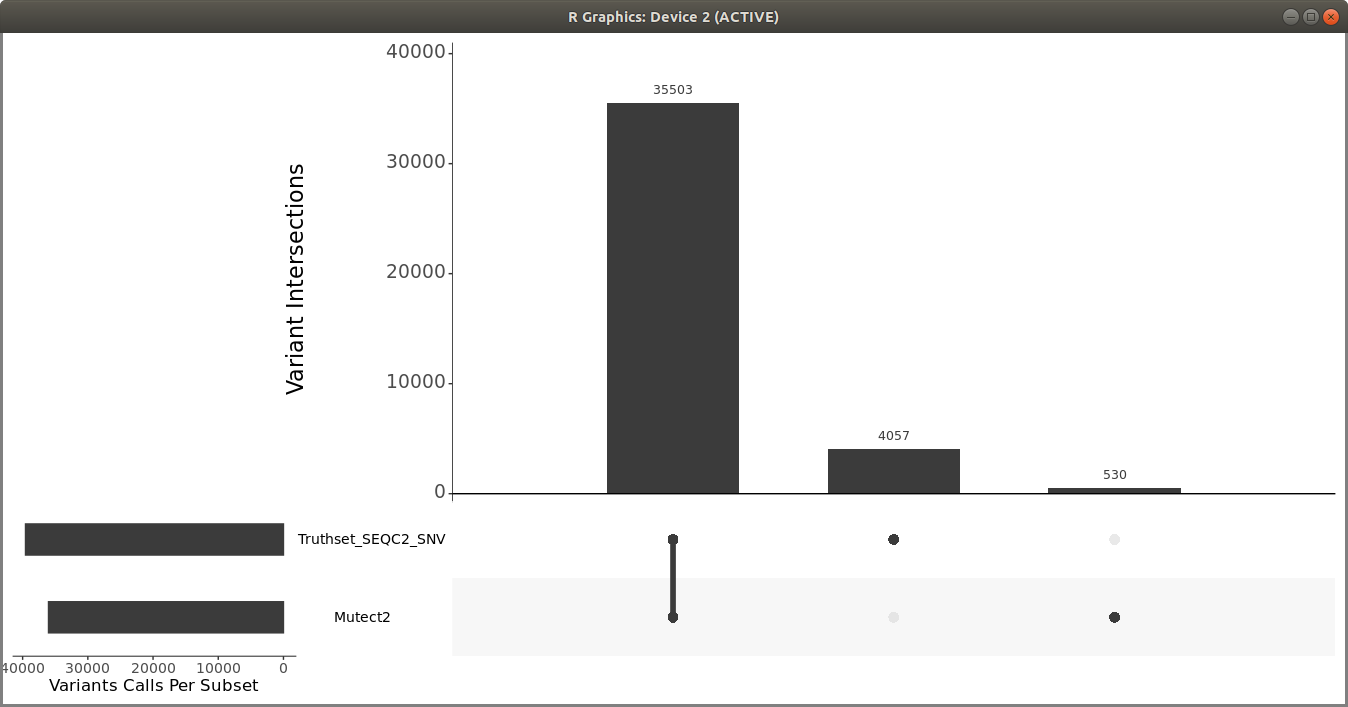
Once you have the filtered Mutect2 calls, use R vcfR and upSetR packages to generate an upset plot to evaluate the performance of the variant calling pipeline. Use the following code to generate an upset plot:
> library(vcfR)
> library(UpSetR)
> ## SEQC2 Somatic SNV upset plot
> ## Load the vcf files from truthset and Mutect2
> Truthset_snv_hc <- read.vcfR("high-confidence_sSNV_in_HC_regions_v1.2.hc.vcf.gz")
> mutect_snv_hc <- read.vcfR("SRR7890824-SRR7890827-WGS_FD.SNV-MNV.FilterMutectCalls.hc.PASS.vcf")
> ### extract chromosome and position to compare
> Truthset_snv_hc <-
as.vector(paste(Truthset_snv_hc@fix[, "CHROM"], Truthset_snv_hc@fix[, "POS"], sep = "_"))
> mutect_snv_hc <-
as.vector(paste(mutect_snv_hc@fix[, "CHROM"], mutect_snv_hc@fix[, "POS"], sep = "_"))
> ## create the read set
> read_sets <- list(Truthset_SEQC2_SNV = Truthset_snv_hc,
Mutect2 = mutect_snv_hc)
> upset(fromList(read_sets), order.by = c("freq"), keep.order = TRUE, sets = c("Mutect2", "Truthset_SEQC2_SNV"), point.size = 3.5, line.size = 2, mainbar.y.label = "Variant Intersections", text.scale = c(2, 2, 1.5, 1.5, 1.5, 1.5),sets.x.label = "Variants Calls Per Subset")
You should get a plot similar to the following:
You can also use som.py from the Illumina hap.py package to compare the two VCF files.
On their own, the VCF files produced by DeepVariant, HaplotypeCaller, and Strelka are not particularly useful. You’ll need to download all of the databases and indices before you can annotate the VCF with them. This how-to uses the following databases:
1000Genomes: a database of variants called in 2,504 individuals from healthy individuals.
ClinVar: an NCBI database of variants associated with disease.
gnomAD: The Genome Aggregation Database contains population variants from 125,748 exomes and 15,708 whole genomes.
COSMIC: Coding and non-coding variants from the Catalogue of Somatic Mutations in Cancer
dbSNP: SNPs, MNPs and indels for both common and clinical mutations
Downloading the COSMIC VCF files requires signing in to the COSMIC site; instructions for doing so are not included here. Make sure to use the normalized variant files; otherwise, variant alleles at indels may not match between the database and query. For this how-to, you will annotate the query VCF(s) with both the COSMIC NonCoding and COSMIC Coding files.
https://www.nvidia.com/en-us/clara/genomics/
https://sites.google.com/view/seqc2/home
https://gatk.broadinstitute.org/hc/en-us/articles/360036360312-Mutect2
https://github.com/Illumina/strelka/blob/v2.9.x/docs/userGuide/README.md
https://bioinformatics.mdanderson.org/public-software/muse/
https://csb5.github.io/lofreq/
https://github.com/genome/somatic-sniper/blob/master/gmt/documentation.md
https://github.com/Illumina/manta
https://github.com/Illumina/hap.py
https://cran.r-project.org/web/packages/vcfR/readme/README.html
https://cran.r-project.org/web/packages/UpSetR/vignettes/basic.usage.html