somatic (Somatic Variant Caller)
Run a somatic variant workflow.
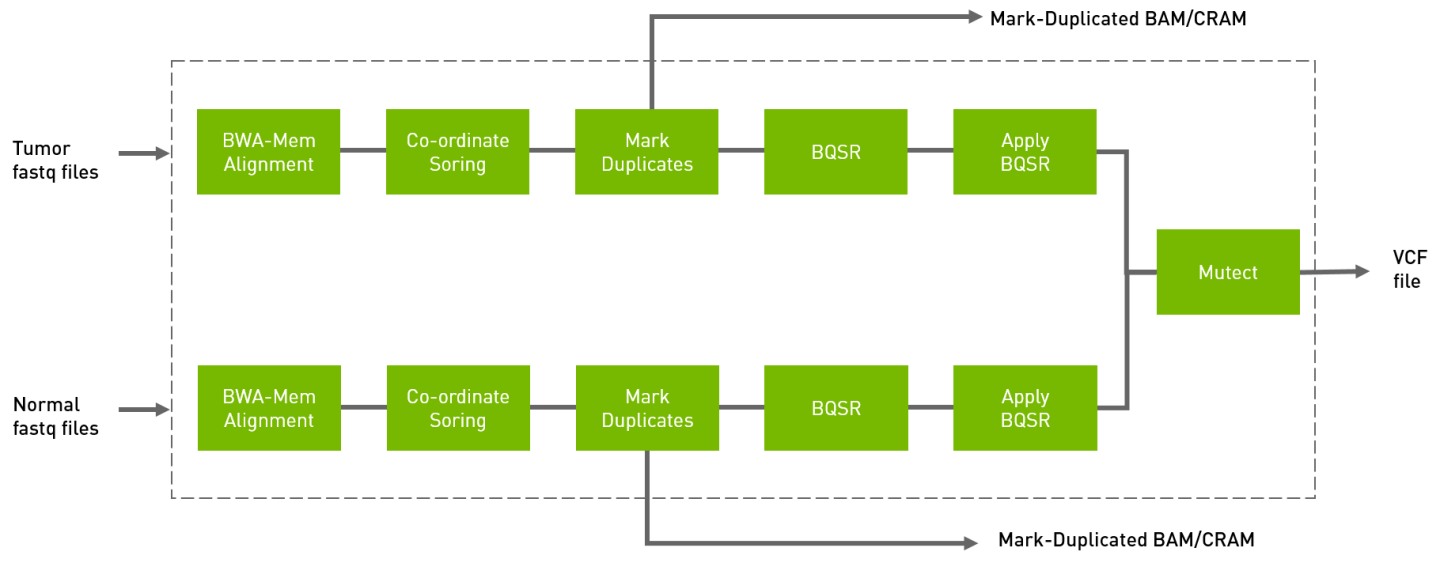
The somatic tool processes the tumor FASTQ files, and optionally normal FASTQ files and knownSites files, and generates tumor or tumor/normal analysis. The output is in VCF format.
Internally the somatic tool runs several other Parabricks tools, thereby simplifying your work flow.
# The command line below will run tumor-only analysis.
# This command assumes all the inputs are in INPUT_DIR and all the outputs go to OUTPUT_DIR.
$ docker run --rm --gpus all --volume INPUT_DIR:/workdir --volume OUTPUT_DIR:/outputdir \
--workdir /workdir \
nvcr.io/nvidia/clara/clara-parabricks:4.1.0-1 \
pbrun somatic \
--ref /workdir/${REFERENCE_FILE} \
--in-tumor-fq /workdir/${INPUT_FASTQ_1} /workdir/${INPUT_FASTQ_2} \
--bwa-options="-Y" \
--out-vcf /outputdir/${OUTPUT_VCF} \
--out-tumor-bam /outputdir/${OUTPUT_BAM}
# The command line below will run tumor-normal analysis.
# This command assumes all the inputs are in INPUT_DIR and all the outputs go to OUTPUT_DIR.
$ docker run --rm --gpus all --volume INPUT_DIR:/workdir --volume OUTPUT_DIR:/outputdir \
--workdir /workdir \
nvcr.io/nvidia/clara/clara-parabricks:4.1.0-1 \
pbrun somatic \
--ref /workdir/${REFERENCE_FILE} \
--knownSites /workdir/${KNOWN_SITES_FILE} \
--in-tumor-fq /workdir/${INPUT_TUMOR_FASTQ_1} /workdir/${INPUT_TUMOR_FASTQ_2} "@RG\tID:sm_tumor_rg1\tLB:lib1\tPL:bar\tSM:sm_tumor\tPU:sm_tumor_rg1" \
--bwa-options="-Y" \
--out-vcf /outputdir/${OUTPUT_VCF} \
--out-tumor-bam /outputdir/${OUTPUT_TUMOR_BAM} \
--out-tumor-recal-file /outputdir/${OUTPUT_RECAL_FILE} \
--in-normal-fq /workdir/${INPUT_NORMAL_FASTQ_1} /workdir/${INPUT_NORMAL_FASTQ_2} "@RG\tID:sm_normal_rg1\tLB:lib1\tPL:bar\tSM:sm_normal\tPU:sm_normal_rg1" \
--out-normal-bam /outputdir/${OUTPUT_NORMAL_BAM}
# The commands below will run tumor-normal analysis.
#
# Run bwa mem on the tumor FASTQ files then sort the BAM by coordinates.
$ bwa mem \
-t 32 \
-K 10000000 \
-Y \
-R '@RG\tID:sample_rg1\tLB:lib1\tPL:bar\tSM:sample\tPU:sample_rg1' \
${REFERENCE_FILE} ${TUMOR_FASTQ_1} ${TUMOR_FASTQ_2} | \
gatk SortSam \
--java-options -Xmx30g \
--MAX_RECORDS_IN_RAM 5000000 \
-I /dev/stdin \
-O tumor_cpu.bam \
--SORT_ORDER coordinate
# Mark duplicates.
$ gatk MarkDuplicates \
--java-options -Xmx30g \
-I tumor_cpu.bam \
-O tumor_mark_dups_cpu.bam \
-M tumor_metrics.txt
# Generate a BQSR report.
$ gatk BaseRecalibrator \
--java-options -Xmx30g \
--input tumor_mark_dups_cpu.bam \
--output ${OUTPUT_TUMOR_RECAL_FILE} \
--known-sites ${KNOWN_SITES_FILE} \
--reference ${REFERENCE_FILE}
# Apply the BQSR report.
$ gatk ApplyBQSR \
--java-options -Xmx30g \
-R ${REFERENCE_FILE} \
-I tumor_cpu.bam \
--bqsr-recal-file ${TUMOR_OUTPUT_RECAL_FILE} \
-O ${OUTPUT_TUMOR_BAM}
# Now repeat all the above steps, only with the normal FASTQ data.
$ bwa mem \
-t 32 \
-K 10000000 \
-Y \
-R '@RG\tID:sample_rg1\tLB:lib1\tPL:bar\tSM:sample\tPU:sample_rg1' \
${REFERENCE_FILE} ${NORMAL_FASTQ_1} ${NORMAL_FASTQ_2} | \
gatk SortSam \
--java-options -Xmx30g \
--MAX_RECORDS_IN_RAM 5000000 \
-I /dev/stdin \
-O normal_cpu.bam \
--SORT_ORDER coordinate
# Mark duplicates.
$ gatk MarkDuplicates \
--java-options -Xmx30g \
-I normal_cpu.bam \
-O normal_mark_dups_cpu.bam \
-M normal_metrics.txt
# Generate a BQSR report.
$ gatk BaseRecalibrator \
--java-options -Xmx30g \
--input normal_mark_dups_cpu.bam \
--output ${OUTPUT_NORMAL_RECAL_FILE} \
--known-sites ${KNOWN_SITES_FILE} \
--reference ${REFERENCE_FILE}
# Apply the BQSR report.
$ gatk ApplyBQSR \
--java-options -Xmx30g \
-R ${REFERENCE_FILE} \
-I normal_cpu.bam \
--bqsr-recal-file ${OUTPUT_NORMAL_RECAL_FILE} \
-O ${OUTPUT_NORMAL_BAM}
# Finally, run Mutect2 on the normal and tumor data.
$ gatk Mutect2 \
-R ${REFERENCE_FILE} \
--input ${OUTPUT_TUMOR_BAM} \
--tumor-sample tumor \
--input ${OUTPUT_NORMAL_BAM} \
--normal-sample normal \
--output ${OUTPUT_VCF}
Run the tumor normal somatic pipeline from FASTQ to VCF.
Input/Output file options
- --ref REF
- --in-tumor-fq [IN_TUMOR_FQ [IN_TUMOR_FQ ...]]
- --in-se-tumor-fq [IN_SE_TUMOR_FQ [IN_SE_TUMOR_FQ ...]]
- --in-normal-fq [IN_NORMAL_FQ [IN_NORMAL_FQ ...]]
- --in-se-normal-fq [IN_SE_NORMAL_FQ [IN_SE_NORMAL_FQ ...]]
- --knownSites KNOWNSITES
- --interval-file INTERVAL_FILE
- --out-vcf OUT_VCF
- --out-tumor-bam OUT_TUMOR_BAM
- --out-normal-bam OUT_NORMAL_BAM
- --out-tumor-recal-file OUT_TUMOR_RECAL_FILE
- --out-normal-recal-file OUT_NORMAL_RECAL_FILE
Path to the reference file. (default: None)
Option is required.
Path to the pair-ended FASTQ files followed by optional read group with quotes (Example: "@RGtID:footLB:lib1tPL:bartSM:20"). The files can be in fastq or fastq.gz format. Either all sets of inputs have a read group, or none should have one, and it will be automatically added by the pipeline. This option can be repeated multiple times. Example 1: --in-tumor-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz --in-tumor-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz. Example 2: --in-tumor-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz "@RG ID:footLB:lib1tPL:bartSM:sm_tumortPU:unit1" --in-tumor-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz "@RG ID:foo2tLB:lib1tPL:bartSM:sm_tumortPU:unit2". For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU. (default: None)
Path to the single-ended FASTQ file followed by an optional read group with quotes (Example: "@RGtID:footLB:lib1tPL:bartSM:sampletPU:foo"). The file must be in fastq or fastq.gz format. Either all sets of inputs have a read group, or none should have one; if no read group is provided, one will be added automatically by the pipeline. This option can be repeated multiple times. Example 1: --in-se-tumor-fq sampleX_1.fastq.gz --in-se-tumor-fq sampleX_2.fastq.gz . Example 2: --in-se-tumor-fq sampleX_1.fastq.gz "@RGtID:footLB:lib1tPL:bartSM:tumortPU:unit1" --in-se-tumor-fq sampleX_2.fastq.gz "@RGtID:foo2tLB:lib1tPL:bartSM:tumortPU:unit2" . For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU. (default: None)
Path to the pair-ended FASTQ files followed by an optional read group with quotes (Example: "@RGtID:footLB:lib1tPL:bartSM:20"). The files must be in fastq or fastq.gz format. Either all sets of inputs have a read group, or none should have one; if no read group is provided, one will be automatically added by the pipeline. This option can be repeated multiple times. Example 1: --in-normal-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz --in-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz . Example 2: --in-normal-fq sampleX_1_1.fastq.gz sampleX_1_2.fastq.gz "@RG ID:footLB:lib1tPL:bartSM:sm_normaltPU:unit1" --in-normal-fq sampleX_2_1.fastq.gz sampleX_2_2.fastq.gz "@RG ID:foo2tLB:lib1tPL:bartSM:sm_normaltPU:unit2". For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU. (default: None)
Path to the single-ended FASTQ file followed by optional read group with quotes (Example: "@RGtID:footLB:lib1tPL:bartSM:sampletPU:foo"). The file must be in fastq or fastq.gz format. Either all sets of inputs have a read group, or none should have one; if no read group is provided, one will be added automatically by the pipeline. This option can be repeated multiple times. Example 1: --in-se-normal-fq sampleX_1.fastq.gz --in-se-normal-fq sampleX_2.fastq.gz . Example 2: --in-se-normal-fq sampleX_1.fastq.gz "@RGtID:footLB:lib1tPL:bartSM:normaltPU:unit1" --in-se-normal-fq sampleX_2.fastq.gz "@RGtID:foo2tLB:lib1tPL:bartSM:normaltPU:unit2" . For the same sample, Read Groups should have the same sample name (SM) and a different ID and PU. (default: None)
Path to a known indels file. The file must be in vcf.gz format. This option can be used multiple times. (default: None)
Path to an interval file in one of these formats: Picard-style (.interval_list or .picard), GATK-style (.list or .intervals), or BED file (.bed). This option can be used multiple times. (default: None)
Path of the VCF file after Variant Calling. (default: None)
Option is required.
Path of the BAM file for tumor reads. (default: None)
Option is required.
Path of the BAM file for normal reads. (default: None)
Path of the report file after Base Quality Score Recalibration for tumor sample. (default: None)
Path of the report file after Base Quality Score Recalibration for normal sample. (default: None)
Tool Options:
- -L INTERVAL, --interval INTERVAL
- --bwa-options BWA_OPTIONS
- --no-warnings
- --gpuwrite
- --gpusort
- --low-memory
- --filter-flag FILTER_FLAG
- --skip-multiple-hits
- --min-read-length MIN_READ_LENGTH
- --align-only
- --no-markdups
- --fix-mate
- --markdups-assume-sortorder-queryname
- --markdups-picard-version-2182
- --optical-duplicate-pixel-distance OPTICAL_DUPLICATE_PIXEL_DISTANCE
- -ip INTERVAL_PADDING, --interval-padding INTERVAL_PADDING
- --ploidy PLOIDY
- --max-mnp-distance MAX_MNP_DISTANCE
- --mutectcaller-options MUTECTCALLER_OPTIONS
- --tumor-read-group-sm TUMOR_READ_GROUP_SM
- --tumor-read-group-lb TUMOR_READ_GROUP_LB
- --tumor-read-group-pl TUMOR_READ_GROUP_PL
- --tumor-read-group-id-prefix TUMOR_READ_GROUP_ID_PREFIX
- --normal-read-group-sm NORMAL_READ_GROUP_SM
- --normal-read-group-lb NORMAL_READ_GROUP_LB
- --normal-read-group-pl NORMAL_READ_GROUP_PL
- --normal-read-group-id-prefix NORMAL_READ_GROUP_ID_PREFIX
Interval within which to call bqsr from the input reads. All intervals will have a padding of 100 to get read records, and overlapping intervals will be combined. Interval files should be passed using the --interval-file option. This option can be used multiple times (e.g. "-L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000"). (default: None)
Pass supported bwa mem options as one string. The current original bwa mem supported options are -M, -Y and -T (e.g. --bwa-options="-M -Y") (default: None)
Suppress warning messages about system thread and memory usage. (default: None)
Use one GPU to accelerate writing final BAM. (default: None)
Use GPUs to accelerate sorting and marking. (default: None)
Use low memory mode (default: None)
Don't generate SAM entries in the output if the entry's flag's meet this criteria. Criteria: (flag & filter != 0) (default: 0)
Filter SAM entries whose length of SA is not 0 (default: None)
Skip reads below minimum read length. They will not be part of output (default: None)
Generate output BAM after bwa-mem. The output will not be co-ordinate sorted or duplicates will not be marked (default: None)
Do not perform the Mark Duplicates step. Return BAM after sorting. (default: None)
Add mate cigar (MC) and mate quality (MQ) tags to the output file. (default: None)
Assume the reads are sorted by queryname for Marking Duplicates. This will mark secondary, supplementary, and unmapped reads as duplicates as well. This flag will not impact variant calling while increasing processing times. (default: None)
Assume marking duplicates to be similar to Picard version 2.18.2. (default: None)
The maximum offset between two duplicate clusters in order to consider them optical duplicates. Ignored if --out-duplicate-metrics is not passed. (default: None)
Amount of padding (in base pairs) to add to each interval you are including. (default: None)
Ploidy assumed for the BAM file. Currently only haploid (ploidy 1) and diploid (ploidy 2) are supported. (default: 2)
Two or more phased substitutions separated by this distance or less are merged into MNPs. (default: 1)
Pass supported mutectcaller options as one string. The following are currently supported original mutectcaller options: -pcr-indel-model <NONE, HOSTILE, AGGRESSIVE, CONSERVATIVE> (e.g. --mutectcaller-options="-pcr-indel-model HOSTILE"). (default: None)
SM tag for read groups for tumor sample. (default: None)
LB tag for read groups for tumor sample. (default: None)
PL tag for read groups for tumor sample. (default: None)
Prefix for ID and PU tag for read groups for tumor sample. This prefix will be used for all pair of tumor FASTQ files in this run. The ID and PU tag will consist of this prefix and an identifier which will be unique for a pair of FASTQ files. (default: None)
SM tag for read groups for normal sample. (default: None)
LB tag for read groups for normal sample. (default: None)
PL tag for read groups for normal sample. (default: None)
Prefix for ID and PU tags for read groups of a normal sample. This prefix will be used for all pairs of normal FASTQ files in this run. The ID and PU tags will consist of this prefix and an identifier that will be unique for a pair of FASTQ files. (default: None)
Common options:
- --logfile LOGFILE
- --tmp-dir TMP_DIR
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --no-seccomp-override
- --version
Path to the log file. If not specified, messages will only be written to the standard error output. (default: None)
Full path to the directory where temporary files will be stored.
Full path to the PetaGene installation directory. By default, this should have been installed at /opt/petagene. Use of this option also requires that the PetaLink library has been preloaded by setting the LD_PRELOAD environment variable. Optionally set the PETASUITE_REFPATH and PGCLOUD_CREDPATH environment variables that are used for data and credentials (default: None)
Do not delete the directory storing temporary files after completion.
Do not override seccomp options for docker (default: None).
View compatible software versions.
GPU options:
- --num-gpus NUM_GPUS
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used.