Benchmarking Whole Genome and Whole Exome Data from Complete Genomics using Parabricks on AWS
This is a quick start guide for benchmarking Parabricks germline workflows using data from Complete Genomics sequencers. Parabricks is a GPU accelerated toolkit for secondary analysis in genomics. In this guide, we will show that Parabricks runs in a fast, and therefore cost effective, manner on the cloud using data from the DNBSEQ-T7 and DNBSEQ-G400 sequencers from Complete Genomics.
Genomic files such as FASTQ and BAM files can easily reach into the hundreds of GB each. When running studies that involve hundreds of thousands of these files, it easily becomes terabytes of data and processing all of that data becomes very costly. This is especially apparent when running on the cloud where users are charged by the hour, so every minute of compute counts. The faster we can churn through this data, the lower the cost will be.
The code that accompanies this example can be found on GitHub.
Software
These benchmarks were performed using Parabricks version 4.0.1-1 which is publicly available as a Docker container on the NVIDIA GPU Cloud (NGC) by running the following command:
docker pull nvcr.io/nvidia/clara/clara-parabricks:4.0.1-1
Other software prerequisites include:
Software |
Version |
Purpose |
|---|---|---|
| bwa | 0.7.18 | Indexing the reference |
| seqtk | 1.4 | Downsampling FASTQ |
To maximize Parabricks performance, it’s best that all the file reading and writing happen on the fast SSD on the machine. To find the path of the SSD, run:
lsblk
Export the mount point as the environment variable NVME_DRIVE so that the scripts in this repository know where to download and run the data from:
export NVME_DRIVE=/opt/dlami/nvme
Hardware
For Parabricks, there are two categories of GPUs that we recommend: High Performance GPUs (A100, H100, L40S) and Low Cost GPUs (A10, L4). These benchmarks were validated using L40S instances and L4 instances on AWS, however any similarly configured machine will work. Be sure to check the Parabricks documentation for minimum requirements. Below are the exact configurations used in our validation:
Configuration |
L4 |
L40S |
|---|---|---|
| Instance Type | g6.24xlarge | g6e.24xlarge |
| OS | Ubuntu | Ubuntu |
| AMI | Deep Learning Nvidia Driver | Deep Learning Nvidia Driver |
| Num GPUs | 4 | 4 |
| vCPUs | 96 | 96 |
| CPU Memory | 384 GB | 768 GB |
| On Demand Cost per Hour* | $6.68 | $15.07 |
For these benchmarks we will use NA12878 whole genome (WGS) data from the DNBSEQ-T7 and DNBSEQ-G400 Complete Genomics sequencers. All the data including the FASTQ, reference, and other accessory files are hosted publicly and can be downloaded using:
download.sh
Preprocessing
The data as it exists publicly is almost ready to use for our benchmarking. For an apples-to-apples comparison, we want both of the WGS samples to have the same coverage. The T7 WGS data has a coverage of 46x and the G400 WGS data has a coverage of 30x. To resolve this, we will downsample the T7 WGS data by 65%. To achieve this, we can run:
downsample.sh 0.65
This will rename E100030471QC960_L01_48_1.fq.gz to E100030471QC960_L01_48_1.30x.fq.gz and then the data will be ready to run through the benchmarks.
The resulting coverages, numbers of reads, and file sizes are summarized for each sample in the table below:
T7 |
G400 |
|
|---|---|---|
| 30x | 30x | |
| 658 Million | 955 Million | |
| 72 GB | 69 GB |
Looking in the benchmarks folder will show us what benchmarking scripts are available:
benchmarks/
├── L4
│ ├── deepvariant.sh
│ └── germline.sh
└── L40S
├── deepvariant.sh
└── germline.sh
For each set of hardware, there is a germline and a deepvariant script. The separation is due to different optimization flags used for each configuration. To learn more about these optimizations, check out the Parabricks documentation . The germline script runs Parabricks germline pipeline, which aligns the FASTQ files and runs HaplotypeCaller. The deepvariant script runs the Parabricks deepvariant variant caller.
The benchmark.sh script accepts one argument for the hardware type, which matches the folder name within the benchmarks folder. For example, to run the L4 benchmarks, we can run:
./benchmarks L4
and similarly to run the L40S benchmarks, we can run:
./benchmarks L40S
The Parabricks software outputs for how long each step of the pipelines ran and it is these numbers that we have recorded in the tables below. After running the benchmarks, the runtimes will be captured in log files located at ${NVME_DIR}/data/logs. Below is a graph of the runtimes for each sample on each GPU type.
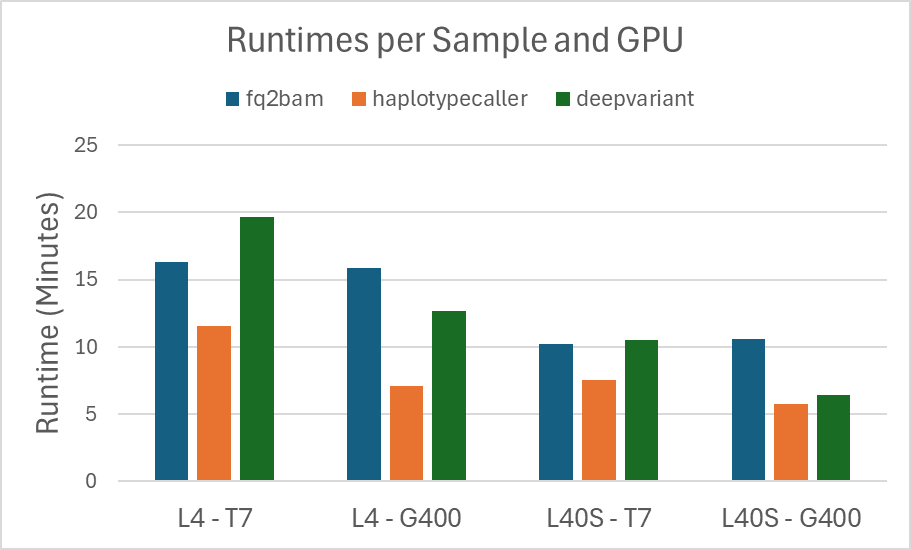
Figure 1. Runtimes for each sample on each GPU type for fq2bam, haplotypecaller and deepvariant.
As we expect, the runtime for the L40S instance is faster than the runtimes for the L4 instance, for each sample. This difference is reflected in the cost as well, shown in the graph below:
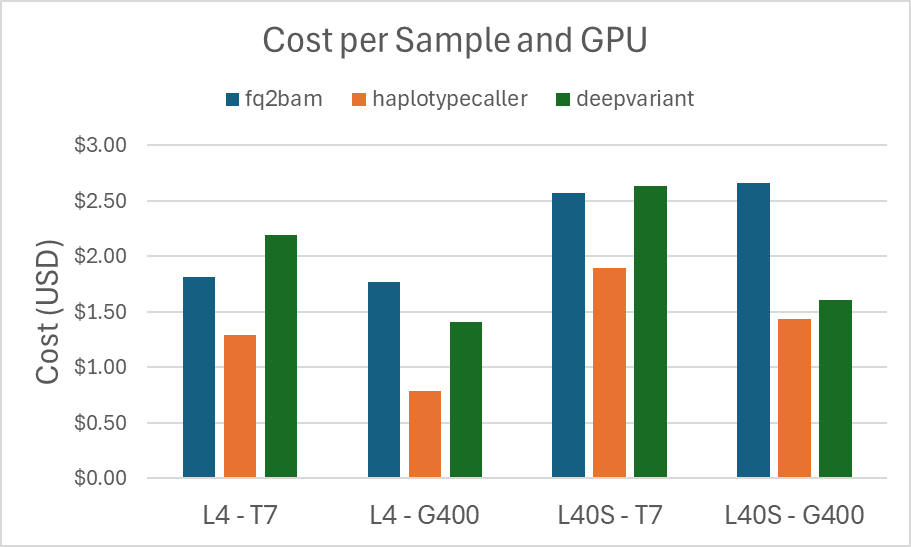
Figure 2. Costs for each sample on each GPU type for fq2bam, haplotypecaller, and deepvariant.
The exact numbers used for these graphs are compiled together in the table below.
L4 |
L40S |
|||||||
|---|---|---|---|---|---|---|---|---|
T7 |
G400 |
T7 |
G400 |
|||||
Runtime |
Cost |
Runtime |
Cost |
Runtime |
Cost |
Runtime |
Cost |
|
| fq2bam | 17 | $1.85 | 16 | $1.77 | 10 | $2.39 | 11 | $2.66 |
| haplotypecaller | 7 | $0.82 | 7 | $0.68 | 6 | $1.44 | 6 | $1.44 |
| deepvariant | 13 | $1.44 | 13 | $1.41 | 7 | $1.70 | 6 | $1.60 |
Aside from the runtime numbers, it’s important to check that the quality of the variants matches closely with the truth set.
The NA12878 ground truth VCF can be found on the NIH FTP. Since this VCF was run using the GRCh37 reference but our samples were run using UCSC hg19 reference, we first need to do a liftover and then we can run concordance.
As an optional step, this can be done using the liftover.sh and
concordance.sh scripts respectively.
Below is a table of the results that we achieved using this workflow:
DeepVariant Concordance |
|||||||
|---|---|---|---|---|---|---|---|
| Type | Filter | TRUTH.TP | TRUTH.FN | QUERY.FP | METRIC.Recall | METRIC.Precision | METRIC_F1_Score |
| INDEL | ALL | 460832 | 6052 | 1389 | 0.987037 | 0.997107 | 0.992047 |
| INDEL | PASS | 460832 | 6052 | 1389 | 0.987037 | 0.997107 | 0.992047 |
| SNP | ALL | 3213043 | 38814 | 6106 | 0.988064 | 0.998104 | 0.993059 |
| SNP | PASS | 3213043 | 38814 | 6106 | 0.988064 | 0.998104 | 0.993059 |
DeepVariant Concordance |
|||||||
|---|---|---|---|---|---|---|---|
| Type | Filter | TRUTH.TP | TRUTH.FN | QUERY.FP | METRIC.Recall | METRIC.Precision | METRIC_F1_Score |
| INDEL | ALL | 460832 | 6052 | 1389 | 0.987037 | 0.997107 | 0.992047 |
| INDEL | PASS | 460832 | 6052 | 1389 | 0.987037 | 0.997107 | 0.992047 |
| SNP | ALL | 3213043 | 38814 | 6106 | 0.988064 | 0.998104 | 0.993059 |
| SNP | PASS | 3213043 | 38814 | 6106 | 0.988064 | 0.998104 | 0.993059 |
DeepVariant Concordance |
|||||||
|---|---|---|---|---|---|---|---|
| Type | Filter | TRUTH.TP | TRUTH.FN | QUERY.FP | METRIC.Recall | METRIC.Precision | METRIC_F1_Score |
| INDEL | ALL | 459953 | 6931 | 2320 | 0.985155 | 0.995165 | 0.990135 |
| INDEL | PASS | 459953 | 6931 | 2320 | 0.985155 | 0.995165 | 0.990135 |
| SNP | ALL | 3208100 | 43757 | 7435 | 0.986544 | 0.997689 | 0.992085 |
| SNP | PASS | 3208100 | 43757 | 7435 | 0.986544 | 0.997689 | 0.992085 |
DeepVariant Concordance |
|||||||
|---|---|---|---|---|---|---|---|
| Type | Filter | TRUTH.TP | TRUTH.FN | QUERY.FP | METRIC.Recall | METRIC.Precision | METRIC_F1_Score |
| INDEL | ALL | 459224 | 7660 | 5812 | 0.983593 | 0.987961 | 0.985772 |
| INDEL | PASS | 459224 | 7660 | 5812 | 0.983593 | 0.987961 | 0.985772 |
| SNP | ALL | 3199856 | 52001 | 47560 | 0.984009 | 0.985358 | 0.984683 |
| SNP | PASS | 3199856 | 52001 | 47560 | 0.984009 | 0.985358 | 0.984683 |
Just as we expect with Parabricks, we are seeing Precision and F1 scores upwards of 99%, confirming that the variant callers are accurate with respect to the ground truth.
In this guide, we showed how to download WGS data from Complete Genomics, run it through alignment (fq2bam) and variant calling (haplotypecaller and deepvariant) on AWS, show the runtime and total cost per sample, and finally demonstrated the concordance results against a truth set. Combined, this shows that Parabricks supports data from Complete Genomics sequencers, in that it runs quickly and accurately on the tools.