

fq2bam_meth
fq2bam_meth
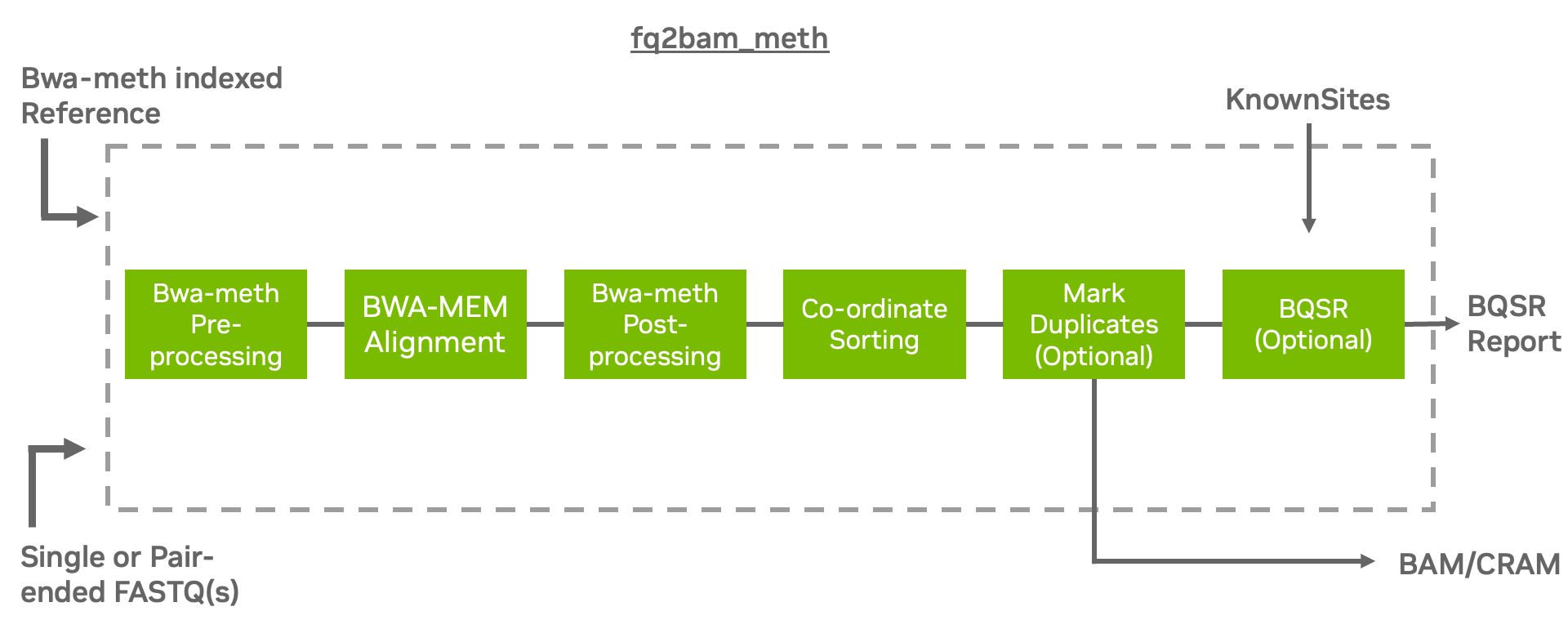
Generate BAM/CRAM output given one or more pairs of FASTQ files from bisulfite sequencing (BS-Seq). Can also optionally generate a BQSR report.
Refer to the fq2bam_meth Reference section for a detailed listing of all available options.
What is fq2bam_meth?
The tool fq2bam_meth is a fast, accurate algorithm for mapping methylated DNA sequence reads to a reference genome, performing local alignment, and producing alignment for different parts of the query sequence. It implements the baseline tool bwa-meth [^note] [^note] in a performant method using fq2bam (BWA-MEM + GATK) as a backend for processing on GPU.
Why fq2bam_meth?
fq2bam_meth is the Parabricks wrapper for bwa-meth, which will sort the output and can mark duplicates and recalibrate base quality scores in line with GATK best practices.
The Parabricks fq2bam_meth tool is capable of handling longer reads and is less sensitive to errors
than other alignment algorithms. We enable fast and accurate whole-genome bisulfite sequencing (WGBS)
to detect DNA-methylation at the single base pair level [^note].
Some of the advantages of using fq2bam_meth over similar tools include:
- It is faster than many other BS-Seq alignment algorithms, making it the ideal choice for high-throughput analysis.
- It maintains compatibility with existing CPU-based tools.
How Should I Use fq2bam_meth?
fq2bam_meth uses an accelerated version of BWA-MEM to generate BAM/CRAM output
given one or more pairs of FASTQ files from BS-Seq. The user can turn-off marking
of duplicates by adding the --no-markdups option. The BQSR step is only performed if the
--knownSites input and --out-recal-file output options are provided; doing so will also
generate a BQSR report.
Prior to running alignment, the reference genome must be converted using baseline bwa-meth.
The bwa-meth indexing step produces a reference fasta file with a name formatted as fasta.bwameth.c2t.
The indexing preparation step requires running bwameth.py index $REF.fasta.
Baseline bwa-meth requires baseline BWA-MEM to be in the user’s path for indexing functionality.
Note that indexing is a time-consuming prerequisite that should only need to be completed once per reference genome.
The bwameth.py script can be found here.
Quick Start
Useful Options for Performance
Suggested parameters to control host memory use:
- The parameter
--bwa-normalized-queue-capacitycontrols the amount of batches that will be in memory to be processed. By default, the value normalized to the number of GPUs in the run is 10, corresponding to<number of GPUs> * <normalized capacity>for the total queue size. Multiple queues are used. Lowering this value would be a good strategy to reduce host memory use. However, a value that is too low may hamper performance.
Parabricks automatically uses an optimal number of streams based on the GPU’s device memory specifications (by default --bwa-nstreams auto). The user may experiment further with the --bwa-nstreams and --bwa-cpu-thread-pool parameters to potentially achieve better performance.
Additionally, for advanced performance tuning and additional control the option --bwa-primary-cpus allows for more fine-grained control of CPU threading.
Each primary CPU thread drives P CPU thread pool threads as specifed with the option --bwa-cpu-thread-pool. The total number of CPU threads processing the CPU stages of alignment is the product of the --bwa-primary-cpus and --bwa-cpu-thread-pool parameters. This allows the user to control the ratio of “primary” CPU threads, which act indepently, to thread pool threads, which act in unison. Depending on the specific server configuration, input data, and GPUs available, it may be better to switch from more thread pool threads to more primary CPU threads. Changing the number of primary CPU threads may increase the CPU resources required.
-
The parameter
--gpuwriteuses the GPU to compress the final BAM or CRAM file for improved performance during the final stage.--gpuwrite-deflate-algocan be used to control the compression ratio. See below for more details.
-
The parameter
--gpusortuses the GPU to sort the BAM or CRAM file. -
The option
--cigar-on-gpuoffloads CIGAR generation from the CPU (default) to the GPU. Consider enabling it on CPU-constrained systems, or if benchmarking with and without the option indicates the CPU is a bottleneck; it may improve overall runtime.
Compatible CPU-based bwa-meth, GATK4 Commands
The commands below are the bwa-meth-0.2.7, bwa-0.7.15, and GATK4 counterpart of the Parabricks command above. The output from these commands will be identical to the output from the above command. Refer to the Output Comparison page for comparing the results.
Set --bwa-options="-K 10000000" in fq2bam_meth and `-K 10000000 in baseline to produce compatible pair-ended results.
fq2bam_meth will not strip R1 and R2` from read names during preprocessing like baseline bwa-meth.
Source of Mismatches
While Parabricks fq2bam_meth does not lose any accuracy in functionality when compared with BWA-mem
and GATK there are several sources that can result in differences in output files.
- BWA-mem
-Kargument
In pair-ended mode, the chunk size specified by -K can cause small mismatches in the output BAM file.
To get rid of the mismatches here, make sure to pass the same number to both baseline BWA-mem and
Parabricks fq2bam_meth, e.g. -K 10000000.
-
PAaux tag- Parabricks
fq2bam_methputs thePAtag last while BWA-mem puts it first. - BWA-mem rounds
PAtag to 3 digits while Parabricksfq2bam_methdoes not. The aux tag can be filtered by runningsamtools view -x <TAG>
- Parabricks
-
Unmapped reads
- Parabricks
fq2bam_methsorts unmapped reads slightly differently than baseline GATK SortSam. Unmapped reads can be filtered withsamtoolsby doingsamtools view -F 4.
- Parabricks
Options
The —in-fq option takes the names of two FASTQ files, optionally followed by a quoted read group. The FASTQ filenames must not start with a hyphen.
When using the —in-fq-list option a read group is required on each line of the input file.
- Baseline bwa-meth: https://github.com/brentp/bwa-meth/
- Bwa-meth manuscript: http://arxiv.org/abs/1401.1129
- https://doi.org/10.1038/s41587-022-01336-9