DEEPVARIANT GERMLINE PIPELINE
Given one or more pairs of fastq files, you can run the germline variant pipeline workflow to generate output including variants, BAM, and recal.
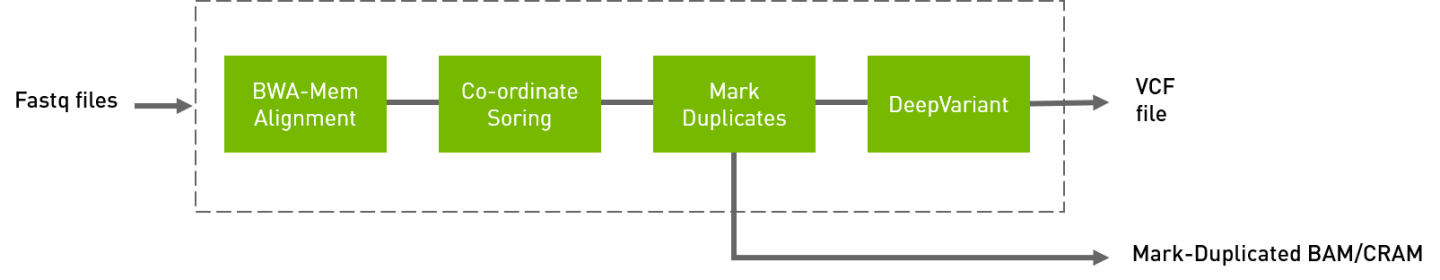
The deepvariant germline pipeline includes alignment, sorting, marking, and deepvariant variant caller. Currently, Deepvariant is supported for T4, V100, and A100 GPUs. The inputs are BWA-indexed reference files, pair-ended fastq files. The outputs of this pipeline are:
Aligned, co-ordinate sorted, duplicated marked bam
Variants in vcf/g.vcf/g.vcf.gz format
- --ref
- --in-fq
- --in-se-fq
- --out-bam
- --out-variants
- --no-markdups
- --out-duplicate-metrics
- --markdups-assume-sortorder-queryname
- --optical-duplicate-pixel-distance
- --read-group-sm
- --read-group-lb
- --read-group-pl
- --read-group-id-prefix
- --interval-file
- --interval
- --gvcf
- --bwa-options
- --disable-use-window-selector-model
- --knownSites KNOWNSITES
- --markdups-picard-version-2182
- --no-warnings
- --optical-duplicate-pixel-distance OPTICAL_DUPLICATE_PIXEL_DISTANCE
- --out-recal-file OUT_RECAL_FILE
- --pb-model-file PB_MODEL_FILE
(required) The reference genome in fasta format. We assume that the indexing required to run bwa has been completed by the user.
(required) Pair ended fastq files. These can be in .fq.gz or .fastq.gz format. You can provide multiple pairs as inputs like so:... --in-fq $fq1 $fq2 --in-fq $fq3 $fq4 ...
(required) Single ended fastq files. These can be in .fastq or .fastq.gz format. You can provide read group information as an optional third argument.
Example 1:
--in-se-fq sampleX.fastq.gz
Example 2:
--in-se-fq sampleX.fastq.gz "@RG\tID:foo\tLB:lib1\tPL:bar\tSM:sample\tPU:unit1"
This option can be repeated multiple times as well.
Example 1:
--in-se-fq sampleX_1.fastq.gz --in-se-fq sampleX_2.fastq.gz
Example 2:
--in-se-fq sampleX_1.fastq.gz "@RG\tID:foo\tLB:lib1\tPL:bar\tSM:sample\tPU:unit1" \
--in-se-fq sampleX_2.fastq.gz "@RG\tID:foo2\tLB:lib1\tPL:bar\tSM:sample\tPU:unit2"
Either all sets of inputs have read group or none should have it and will be automatically added by the pipeline. For the same sample, Read Groups should have the same sample name (SM) and different ID and PU.
(required) Path to the file that will contain BAM output.
(required) Name of VCF/GVCF/GVCF.GZ file after Variant Calling. Absolute or relative path can be given.
Defaults to False.
Do not mark duplicates, generate bam after co-ordinate sorting.
Path of duplicate metrics file after Marking Duplicates.
Assume the reads are sorted by queryname for Marking Duplicates. This will mark secondary, supplementary and unmapped reads as duplicates as well. This flag will not impact variant calling while increasing processing times.
The maximum offset between two duplicate clusters in order to consider them optical duplicates. Ignored if –out-duplicate-metrics is not passed.
SM tag for read groups in this run.
LB tag for read groups in this run.
PL tag for read groups in this run.
Prefix for ID and PU tag for read groups in this run. This prefix will be used for all pair of fastq files in this run. The ID and PU tag will consist of this prefix and an identifier which will be unique for a pair of fastq files.
Path to an interval file for BQSR step with possible formats: Picard-style (.interval_list or .picard), GATK-style (.list or .intervals), or BED file (.bed). This option can be used multiple times (default: None)
(-L) Interval strings within which to call variants from the input reads. Overlapping intervals will be combined. Interval files should be passed using the –interval-file option. This option can be used multiple times. e.g. -L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000 (default: None)
Defaults to False.
Generate variant calls in gVCF format. Final output will have .g.vcf extension.
Pass supported bwa mem options as one string. Current original bwa mem supported options, -M, -Y, -T. e.g. –bwa-options “-M -Y” (default: None)
Change the window selector model from Allele Count Linear to Variant Reads. This option will increase the accuracy and run time (default: Allele Count Linear)
Path to a known indels file. Must be in vcf/vcf.gz format. This option can be used multiple times.
Assume marking duplicates to be similar to Picard version 2.18.2.
Suppress warning messages about system thread and memory usage.
The maximum offset between two duplicate clusters in order to consider them optical duplicates. Ignored if –out-duplicate-metrics is not passed.
Path of report file after Base Quality Score Recalibration. Path can be a Google Cloud Storage object or AWS S3 Storage object.
Path of non-default parabricks model file for deepvariant.
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used. If you are using flexera, please include –gpu-devices too.
Which GPU devices to use for a run. By default, all GPU devices will be used. To use specific GPU devices enter a comma-separated list of GPU device numbers. Possible device numbers can be found by examining the output of the nvidia-smi command. For example, using –gpu-devices 0,1 would only use the first two GPUs.
- --tmp-dir TMP_DIR
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.