VARIANT CALLERS
NVIDIA Clara Parabricks accelerated variant callers
Accelerated bcftools call.
bcftools-call calls variants from mpileup output
QUICK START
$ pbrun bcftoolscall --in-file pileup.bcf \
--out-file output.vcf
COMPATIBLE CPU COMMAND
The command below is the CPU counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
bcftools call pileup.bcf -c -o output.vcf
OPTIONS
- --in-file
- --out-file
- --num-threads
- --variant-sites
Path to the input mpileup file (default: None)
Path of output file. If this option is not used, it will write to standard output (default: None)
Number of threads for worker (default: 1)
Output variant sites only (default: None)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
GPU accelerated haplotypecaller.
This tool runs GPU accelerated haplotypecaller. Users can provide an optional BQSR report to fix the BAM similar to ApplyBQSR. In that case the updated base qualities will be used.

QUICK START
$ pbrun haplotypecaller --ref Ref/Homo_sapiens_assembly38.fasta \
--in-bam mark_dups_gpu.bam \
--in-recal-file recal_gpu.txt \
--out-variants result.vcf
COMPATIBLE GATK4 COMMAND
The command below is the GATK4 counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
# Run ApplyBQSR Step
$ gatk ApplyBQSR --java-options -Xmx30g -R Ref/Homo_sapiens_assembly38.fasta \
-I=mark_dups_cpu.bam --bqsr-recal-file=recal_file.txt -O=cpu_nodups_BQSR.bam
#Run Haplotype Caller
$ gatk HaplotypeCaller --java-options -Xmx30g --input cpu_nodups_BQSR.bam --output \
result_cpu.vcf --reference Ref/Homo_sapiens_assembly38.fasta \
--native-pair-hmm-threads 16
OPTIONS
- --ref
- --in-bam
- --out-variants
- --in-recal-file
- --haplotypecaller-options
- --static-quantized-quals
- --ploidy
- --interval-file
- --interval
- --interval-padding
- --gvcf
- --batch
- --disable-read-filter
- --max-alternate-alleles
- --annotation-group
- --gvcf-gq-bands
- --dont-use-soft-clipped-bases
- --haplotypecaller-options
- --rna
- --read-from-tmp-dir
(required) The reference genome in fasta format.
(required) Path to the input BAM/CRAM file.
(required) Path of .vcf, g.vcf, or g.vcf.gz file.
Path to the input BQSR report. Only required if ApplyBQSR step is needed.
Pass supported haplotype caller options as one string. Current original haplotypecaller supported options: -min-pruning, -standard-min-confidence-threshold-for-calling, -max-reads-per-alignment-start, -min-dangling-branch-length, and -pcr-indel-model .
Use static quantized quality scores to a given number of levels. Repeat this option multiple times for multiple bins.
Defaults to 2.
Ploidy assumed for the bam file. Currently only haploid (ploidy 1) and diploid (ploidy 2) are supported.
Path to an interval file for BQSR step with possible formats: Picard-style (.interval_list or .picard), GATK-style (.list or .intervals), or BED file (.bed). This option can be used multiple times (default: None)
(-L) Interval within which to call variants from the input reads. All intervals will have a padding of 100 to get read records and overlapping intervals will be combined. Interval files should be passed using the –interval-file option. This option can be used multiple times.
e.g. "-L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000" (default: None)
(-ip) Padding size (in base pairs) to add to each interval you are including (default: None)
Defaults to False.
Generate variant calls in gVCF format. When using this option –out-variants file should end with g.vcf or g.vcf.gz. If the --out-variants file ends in gz, the tool will generate gvcf.gz and index for it.
Given an input list of BAMs, run the variant calling of each BAM using one GPU, and process BAMs in parallel based on how many GPUs the system has.
Disable the read filters for bam entries. Currently supported read filters that can be disabled are: MappingQualityAvailableReadFilter, MappingQualityReadFilter, and NotSecondaryAlignmentReadFilter. This option can be repeated multiple times.
Maximum number of alternate alleles to genotype (default: None)
(-G) Which groups of annotations to add to the output variant calls. Currently supported annotation groups: StandardAnnotation, StandardHCAnnotation, AS_StandardAnnotation (default: None)
(-GQB) Exclusive upper bounds for reference confidence GQ bands. Must be in the range [1, 100] and specified in increasing order (default: None)
Dont’ use fot clipped bases for variant calling
Pass supported haplotype caller options as one string. Currently supported original haplotypecaller options:
-min-pruning <int>
-standard-min-confidence-threshold-for-calling <int>
-max-reads-per-alignment-start <int>
-min-dangling-branch-length <int>
-pcr-indel-model <NONE, HOSTILE, AGGRESSIVE, CONSERVATIVE>
e.g. –haplotypecaller-options=”-min-pruning 4 -standard-min-confidence-threshold-for-calling 30”
Run haplotypecaller optimized for RNA Data.
Read from the temporary files generated by fq2bam. Only supported on ampere containers (default: None)
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used. If you are using flexera, please include –gpu-devices too.
Which GPU devices to use for a run. By default, all GPU devices will be used. To use specific GPU devices enter a comma-separated list of GPU device numbers. Possible device numbers can be found by examining the output of the nvidia-smi command. For example, using –gpu-devices 0,1 would only use the first two GPUs.
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
GPU accelerated mutect2.
mutectcaller supports tumor or tumor-normal variant calling. The figure below shows the high level functionality of mutectcaller. All dotted boxes indicate optional data, with some constraints.

QUICK START
$ pbrun mutectcaller --ref Ref/Homo_sapiens_assembly38.fasta \
--in-tumor-bam tumor.bam \
--tumor-name tumor \
--in-normal-bam normal.bam \
--normal-name normal \
--out-vcf output.vcf
COMPATIBLE GATK4 COMMAND
The command below is the GATK4 counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
gatk Mutect2 -R Ref/Homo_sapiens_assembly38.fasta --input tumor.bam --tumor-sample tumor --input normal.bam --normal-sample normal --output output.vcf
OPTIONS
- --ref
- --in-tumor-bam
- --tumor-name
- --out-vcf
- --in-tumor-recal-file
- --in-normal-bam
- --in-normal-recal-file
- --normal-name
- --ploidy
- --interval-file
- --interval
- --interval-padding
- --mutectcaller-options
- --max-mnp-distance
(required) The reference genome in fasta format. We assume that the indexing required to run bwa has been completed by the user.
(required) Path of BAM/CRAM file for tumor reads.
(required) Name of sample for tumor reads.
(required) Path to the VCF output file.
Path of BQSR report for tumor sample.
Path of BAM/CRAM file for normal reads.
Path of BQSR report for normal sample.
Name of sample for normal reads.
Ploidy assumed for the input file. Currently only haploid (ploidy 1) and diploid (ploidy 2) are supported.
Path to an interval file for BQSR step with possible formats: Picard-style (.interval_list or .picard), GATK-style (.list or .intervals), or BED file (.bed). This option can be used multiple times (default: None)
(-L) Interval within which to call variants from the input reads. All intervals will have a padding of 100 to get read records and overlapping intervals will be combined. Interval files should be passed using the --interval-file option. This option can be used multiple times.
e.g. "-L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000" (default: None)
(-ip) Padding size (in base pairs) to add to each interval you are including (default: None)
Pass supported mutectcaller options as one string. Currently supported original mutectcaller options:
-pcr-indel-model <NONE, HOSTILE, AGGRESSIVE, CONSERVATIVE>
e.g. –mutectcaller-options=”-pcr-indel-model HOSTILE” (default: None)
Two or more phased substitutions separated by this distance or less are merged into MNPs. (default: 1)
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used. If you are using flexera, please include –gpu-devices too.
Which GPU devices to use for a run. By default, all GPU devices will be used. To use specific GPU devices enter a comma-separated list of GPU device numbers. Possible device numbers can be found by examining the output of the nvidia-smi command. For example, using –gpu-devices 0,1 would only use the first two GPUs.
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Accelerated Somatic Sniper.
Somatic sniper supports tumor-normal variant calling. Parabricks has Somatic Sniper as a standalone tool or you can use the Somatic Sniper workflow (sniperworkflow) to generate a VCF file from BAM/CRAM.
QUICK START
$ pbrun somaticsniper --ref Ref/Homo_sapiens_assembly38.fasta --min-mapq 1 --no-gain --no-loh --in-tumor-bam tumor.bam --in-normal-bam normal.bam --out-file output.vcf
COMPATIBLE CPU COMMAND
The command below is the CPU counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
bam-somaticsniper -q 1 -G -L -F vcf -f Ref/Homo_sapiens_assembly38.fasta tumor.bam normal.bam output.vcf
OPTIONS
- --ref
- --in-tumor-bam
- --in-normal-bam
- --out-file
- --num-threads
- --min-mapq
- --out-format
- --correct
- --no-gain
- --no-loh
Path to the reference file (default: None)
Path of BAM file for tumor reads. Path can be a Google Cloud Storage object (default: None)(no CRAM support yet)
Path of BAM file for normal reads. Path can be a Google Cloud Storage object (default: None)(no CRAM support yet)
Path of output file (default: None)
Number of threads for worker (default: 1)
Filtering reads with mapping quality less than this value (default: 0)
Type of output format. Possible values are {classic, vcf} (default: classic)
Fix baseline bugs. If this option is not passed, the same output will be generated as baseline (default: None)
Do not report Gain of Reference variants as determined by genotypes (default: None)
Do not report LOH variants as determined by genotypes (default: None)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Somatic sniper workflow to generate VCF from BAM/CRAM input files.
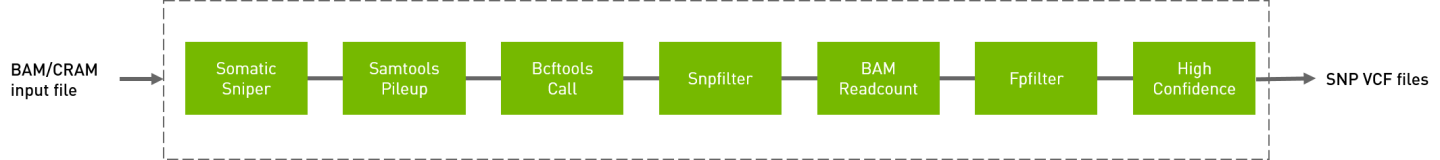
QUICK START
$ pbrun somaticsniper_workflow --ref Ref/Homo_sapiens_assembly38.fasta \
--in-tumor-bam tumor.bam \
--in-normal-bam normal.bam \
--out-prefix output
COMPATIBLE CPU COMMAND
The command below is the CPU counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
bam-somaticsniper -q 1 -G -L -F vcf -f Ref/Homo_sapiens_assembly38.fasta tumor.bam normal.bam output.vcf
bcftools mpileup -A -B -d 2147483647 -Ou -f Ref/Homo_sapiens_assembly38.fasta tumor.bam | bcftools call -c | vcfutils.pl varFilter -Q 20 | awk 'NR > 55 {print}' > output.indel_pileup_Tum.pileup
perl snpfilter.pl --snp-file output.vcf --indel-file output.indel_pileup_Tum.pileup
perl prepare_for_readcount.pl --snp-file output.vcf.SNPfilter
bam-readcount -b 15 -f Ref/Homo_sapiens_assembly38.fasta -l output.vcf.SNPfilter.pos tumor.bam > output.readcounts.rc
perl fpfilter.pl -snp-file output.vcf.SNPfilter -readcount-file output.readcounts.rc
perl highconfidence.pl -snp-file output.vcf.SNPfilter.fp_pass.vcf
OPTIONS
- --ref
- --in-tumor-bam
- --in-normal-bam
- --out-prefix
- --num-threads
- --min-mapq
(required) The reference genome in fasta format. We assume that the indexing required to run bwa has been completed by the user.
(required) Path of BAM file for tumor reads.(no CRAM support yet)
Path of BAM file for normal reads.(no CRAM support yet)
Prefix filename for output data (default: None)
Number of threads for worker (default: 1)
Filtering reads with mapping quality less than this value (default: 1)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Run GPU-accelerated deepvariant algorithm.
Parabricks has accelerated Google Deepvariant to extensively use GPUs and finish 30x WGS analysis in 25 minutes instead of hours. The Parabricks flavor of Deepvariant is more like other command line tools that users are familiar with. It takes the BAM and reference as inputs and produces variants as outputs. Currently, Deepvariant is supported for T4, V100, and A100 GPUs.
QUICK START
$ pbrun deepvariant --ref Ref/Homo_sapiens_assembly38.fasta \
--in-bam mark_dups_gpu.bam \
--out-variants output.vcf
COMPATIBLE GOOGLE DEEPVARIANT COMMANDS
The command below is the Google counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
# Run make_examples in parallel
seq 0 $((N_SHARDS-1)) | \
parallel --eta --halt 2 --joblog "${LOGDIR}/log" --res "${LOGDIR}" \
sudo docker run \
-v ${HOME}:${HOME} \
gcr.io/deepvariant-docker/deepvariant:"${BIN_VERSION}" \
/opt/deepvariant/bin/make_examples \
--mode calling \
--ref "${REF}" \
--reads "${BAM}" \
--examples "${OUTPUT_DIR}/examples.tfrecord@${N_SHARDS}.gz" \
--task {}
# Run call_variants in parallel
sudo docker run \
-v ${HOME}:${HOME} \
gcr.io/deepvariant-docker/deepvariant:"${BIN_VERSION}" \
/opt/deepvariant/bin/call_variants \
--outfile "${CALL_VARIANTS_OUTPUT}" \
--examples "${OUTPUT_DIR}/examples.tfrecord@${N_SHARDS}.gz" \
--checkpoint "${MODEL}"
# Run postprocess_variants in parallel
sudo docker run \
-v ${HOME}:${HOME} \
gcr.io/deepvariant-docker/deepvariant:"${BIN_VERSION}" \
/opt/deepvariant/bin/postprocess_variants \
--ref "${REF}" \
--infile "${CALL_VARIANTS_OUTPUT}" \
--outfile "${FINAL_OUTPUT_VCF}"
OPTIONS
- --ref
- --in-bam
- --out-variants
- --pb-model-file
- --mode
- --proposed-variants
- --interval-file
- --interval
- --disable-use-window-selector-model
- --gvcf
- --norealign-reads
- --sort-by-haplotypes
- --keep-duplicates
- --vsc-min-count-snps
- --vsc-min-count-indels
- --vsc-min-fraction-snps
- --vsc-min-fraction-indels
- --min-mapping-quality
- --min-base-quality
- --alt-aligned-pileup
- --variant-caller
(required) The reference genome in fasta format.
(required) Path to the input BAM/CRAM file.
(required) Name of output VCF file.
Path of a non-default parabricks model file for deepvariant.
Value can be one of [shortread, pacbio, ont]. By default, it is shortread. If mode is set to pacbio, the following defaults are used: –norealign-reads, –alt-aligned-pileup diff_channels, –vsc-min-fraction-indels 0.12. If mode is set to ont, the following defaults are used: -norealign-reads, –variant-caller VCF_CANDIDATE_IMPORTER (default: shortread)
Path of vcf file which has proposed variants for make examples stage (default: None)
Path to an interval file for BQSR step with possible formats: Picard-style (.interval_list or .picard), GATK-style (.list or .intervals), or BED file (.bed). This option can be used multiple times (default: None)
(-L) Interval within which to call variants from the input reads. Overlapping intervals will be combined. Interval files should be passed using the --interval-file option. This option can be used multiple times.
e.g. "-L chr1 -L chr2:10000 -L chr3:20000+ -L chr4:10000-20000" (default: None)
Change the window selector model from Allele Count Linear to Variant Reads. This option will increase the accuracy and run time (default: Allele Count Linear)
Generate variant calls in gVCF format.
Do not locally realign reads before calling variants. Reads longer than 500 bp are never realigned (default: None)
Reads are sorted by haplotypes (using HP tag) (default: None)
Keep reads that are duplicate (default: None)
SNP alleles occurring at least this many times in our AlleleCount will be advanced as candidates (default:2)
Indel alleles occurring at least this many times in our AlleleCount will be advanced as candidates (default: 2)
SNP alleles occurring at least this fraction of all counts in our AlleleCount will be advanced as candidates (default: 0.12)
Indel alleles occurring at least this fraction of all counts in our AlleleCount will be advanced as candidates (default: None)
By default, reads with any mapping quality are kept. Setting this field to a positive integer i will only keep reads that have a MAPQ >= i. Note this only applies to aligned reads (default: 5)
Minimum base quality. This field indicates that we are enforcing a minimum base quality score for alternate alleles. Alternate alleles will only be considered if all bases in the allele have a quality greater than min_base_quality (default: 10)
Value can be one of [none, diff_channels]. Include alignments of reads against each candidate alternate allele in the pileup image. Default is none which turns this feature off (default: None)
Value can be one of [VERY_SENSITIVE_CALLER, VCF_CANDIDATE_IMPORTER]. The caller to use to make examples. If you use VCF_CANDIDATE_IMPORTER, it implies force calling. Default is VERY_SENSITIVE_CALLER
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used. If you are using flexera, please include –gpu-devices too.
Which GPU devices to use for a run. By default, all GPU devices will be used. To use specific GPU devices enter a comma-separated list of GPU device numbers. Possible device numbers can be found by examining the output of the nvidia-smi command. For example, using –gpu-devices 0,1 would only use the first two GPUs.
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
CPU accelerated Copy number variant calling. You need to pass “–extra-tools” to the installer to use this tool.
Run CNVkit with accelerated coverage calculation from read depths.
QUICK START
$ pbrun cnvkit --ref Ref/Homo_sapiens_assembly38.fasta \
--in-bam mark_dups_gpu.bam
--out-file output.vcf
OPTIONS
- --ref
- --in-bam
- --output-dir
- --cnvkit-options
- --generate-vcf
(required) Path to the reference file.
(required) Path to the BAM/CRAM file.
Path to the directory that will contain all of the generated files.
Pass supported cnvkit options as one string. Currently supported options are –count-reads and –drop-low-coverage.
e.g. --cnvkit-options="--count-reads --drop-low-coverage".
Export the output cns to VCF after running batch (default: None)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Call and genotype SVs for short reads using smoove (Original Smoove Project). This tool is not accelerated and original precompiled binary will run on the server.
QUICK START
$ pbrun smoove --ref Ref/Homo_sapiens_assembly38.fasta \
--in-bam in.bam \
--output-dir output \
--name SM
COMPATIBLE CPU COMMAND
The command below is the original CPU counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
./smoove call --fasta Ref/Homo_sapiens_assembly38.fasta \
--outdir output \
--name SM \
in.bam
OPTIONS
- --ref
- --in-bam
- --output-dir
- --name
- --smoove-options
(required) Path to the reference file (default: None)
(required) Path to the input BAM/CRAM file for variant calling (default: None)
(required) Path to the directory that will contain all of the generated files (default: None)
(required) Input sample name (default: None)
Pass supported smoove options as one string. e.g. –smoove-options=”–excludechroms chr4 –noextrafilters” (default: None)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Call variants with accelerated Muse variant caller
QUICK START
$ pbrun muse --ref Ref/Homo_sapiens_assembly38.fasta \
--in-tumor-bam tumor.bam \
--in-normal-bam normal.bam \
--out-vcf out.vcf \
--mode call
COMPATIBLE CPU COMMAND
The command below is the original CPU counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
./MuSE call -f Ref/Homo_sapiens_assembly38.fasta -O out.vcf tumor.bam normal.bam
OPTIONS
- --mode
- --in-tumor-bam
- --in-normal-bam
- --ref
- --in-dbsnp
- --in-callfile
- --out-file
- --out-vcf
- --datatype
- --num-threads
(required) Mode can be call or sump. User should first run muse with call mode and then run with sump mode to generate vcf. Please see MuSE somatic caller documentation for details (default: None)
Path to the input tumor bam file (default: None)
Path to the input normal bam file (default: None)
Path to the input reference file (default: None)
Path to the dbSNP vcf file that should be bgzip compressed, tabix indexed and based on the same reference genome used in call mode (default: None)
Path to the output file of muse call command (default: None)
Output stats file, .MuSE.txt will be automatically appended to this filename (default: None)
Output vcf file (default: None)
Sample-specific error model to use. It can be either E (for whole exomes) or G (for whole genomes) (default: None)
Number of threads for worker. Use 20-24 threads for maximum performance if your system has enough CPU threads (default: 12)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Call variants with high sensitivity, predicting variants below the average base-call quality (Original Lofreq Project). The call part is accelerated.
QUICK START
$ pbrun lofreq --ref Ref/Homo_sapiens_assembly38.fasta \
--in-tumor-bam tumor.bam \
--in-normal-bam normal.bam \
--output-dir output
COMPATIBLE CPU COMMAND
The command below is the original CPU counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
lofreq somatic -n normal.bam -t tumor.bam \
-o output -f /data/Ref/GRCh38.d1.vd1.fa \
--baq-off --no-src-qual --call-rlx-extra-args "@d 2147483647"
OPTIONS
- --ref
- --in-tumor-bam
- --in-normal-bam
- --output-dir
- --in-dbsnp-file
- --ignore-vcf
- --num-threads
- --tumor-mtc
- --tumor-mtc-alpha
- --min-cov MIN_COV
- --germline
- --use-orphan
- --baq-off
- --no-src-qual
(required) Path to the reference file (default: None)
(required) Path of bam file for tumor reads. This option is required (default: None)(no CRAM support yet)
(required) Path of bam file for normal reads. This option is required (default: None)(no CRAM support yet)
(required) Directory for output data (default: None)
Path to an input dbsnp file containing known germline variants. Must be in vcf.gz format with its tabix index (default: None)
Path to an input VCF file containing variants that will be ignored for source quality computation in tumor. If this option is not used, stringently filtered predictions in normal sample will be used by default (default: None)
Number of threads per GPU for each call (default: 4)
Type of multiple testing correction for tumor. Possible values are {bonf,holm-bonf,fdr}. Default value is bonf (default: bonf)
Multiple testing correction alpha for tumor. Default value is 1.000000 (default: 1.0)
Minimum coverage for somatic calls. Default value is 7 (default: 7)
Also list germline calls in separate file (default: None)
Use orphaned/anomalous reads from pairs in all samples (default: None)
Switch use of BAQ off in all samples (default: None)
Disable use of source quality in tumor (default: None)
- --num-gpus NUM_GPUS
- --gpu-devices GPU_DEVICES
Number of GPUs to use for a run. GPUs 0..(NUM_GPUS-1) will be used. If you are using flexera, please include –gpu-devices too.
Which GPU devices to use for a run. By default, all GPU devices will be used. To use specific GPU devices enter a comma-separated list of GPU device numbers. Possible device numbers can be found by examining the output of the nvidia-smi command. For example, using –gpu-devices 0,1 would only use the first two GPUs.
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Structural variant (SV) and indel caller from mapped paired-end sequencing reads. This tool is not accelerated and original precompiled binary will run on the server.
QUICK START
$ pbrun manta --ref Ref/Homo_sapiens_assembly38.fasta \
--in-tumor-bam tumor.bam \
--in-normal-bam normal.bam \
--out-prefix output
OPTIONS
- --ref
- --in-tumor-bam
- --in-normal-bam
- --bed
- --out-prefix
- --num-threads
- --manta-options
Path to the reference file (default: None)
Path of BAM/CRAM file for tumor reads (default: None)
Path of BAM/CRAM file for normal reads. This option can be used multiple times (default: None)
Optional bgzip-compressed/tabix-indexed BED file containing the set of regions to call (default: None)
Prefix filename for output data (default: None)
Number of threads for worker (default: 1)
Pass supported manta options as one string. e.g. –manta-options=”–rna –unstrandedRNA” (default: None)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
SNP and indel caller from mapped paired-end sequencing reads. This tools is not accelerated and original precompiled binary will run on the server.
QUICK START
$ pbrun strelka --ref Ref/Homo_sapiens_assembly38.fasta \
--in-tumor-bam tumor.bam \
--in-normal-bam normal.bam \
--indel-candidates candidates.vcf \
--out-prefix output
OPTIONS
- --ref
- --in-tumor-bam
- --in-normal-bam
- --indel-candidates
- --bed
- --out-prefix
- --num-threads
- --strelka-options
Path to the reference file (default: None)
Path of BAM/CRAM file for tumor reads (default: None)
Path of BAM/CRAM file for normal reads. This option can be used multiple times (default: None)
Path to a VCF of candidate indel alleles. Must be in vcf/vcf.gz format. This option can be used multiple times (default: None)
Optional bgzip-compressed/tabix-indexed BED file containing the set of regions to call (default: None)
Prefix filename for output data (default: None)
Number of threads for worker (default: 1)
Pass supported strelka options as one string. e.g. –strelka-options=”–exome” (default: None)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.
Strelka workflow to generate VCF from BAM/CRAM input files.
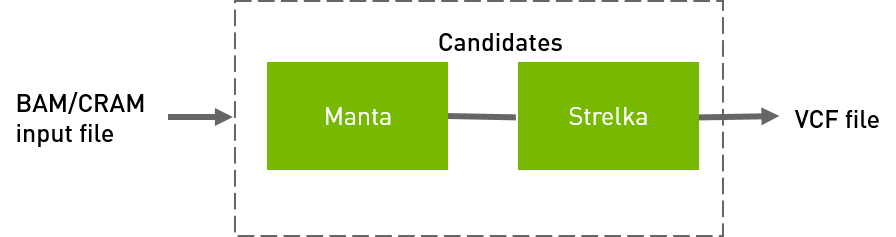
QUICK START
$ pbrun strelka_workflow --ref Ref/Homo_sapiens_assembly38.fasta \
--in-tumor-bam tumor.bam \
--in-normal-bam normal.bam \
--out-prefix output
COMPATIBLE GATK4 COMMAND
The command below is the GATK4 counterpart of the Parabricks command above. The output from these commands will generate the exact same results as the output from the above command. Please look at Output Comparison page on how you can compare the results.
mkdir -p manta_work
python $MANTA_DIR/bin/configManta.py --referenceFasta Ref/Homo_sapiens_assembly38.fasta \
--normalBam normal.bam --tumorBam tumor.bam \
--runDir manta_work
cd manta_work
python ./runWorkflow.py -m local -j ${MAX_NUM_PROCESSORS}
cd ..
mkdir -p strelka_work
python $STRELKA_PATH/configureStrelkaSomaticWorkflow.py \
--referenceFasta Ref/Homo_sapiens_assembly38.fasta \
--normalBam normal.bam --tumorBam tumor.bam \
--indelCandidates ${WORK_PATH}/manta_work/results/variants/candidateSmallIndels.vcf.gz \
--runDir strelka_work
cd strelka_work
python ./runWorkflow.py -m local -j ${MAX_NUM_PROCESSORS}
cd ..
OPTIONS
- --ref
- --in-tumor-bam
- --in-normal-bam
- --out-prefix
- --num-threads
(required) The reference genome in fasta format. We assume that the indexing required to run bwa has been completed by the user.
(required) Path of BAM/CRAM file for tumor reads.
Path of BAM/CRAM file for normal reads.
Prefix filename for output data (default: None)
Number of threads for worker (default: 1)
- --tmp-dir TMP_DIR
- --no-seccomp-override
- --with-petagene-dir WITH_PETAGENE_DIR
- --keep-tmp
- --license-file LICENSE_FILE
- --version
Full path to the directory where temporary files will be stored.
Do not override seccomp options for docker
Full path to the PetaGene installation directory where bin/ and species/ folders are located.
Do not delete the directory storing temporary files after completion.
Path to license file license.bin if not in installation directory.
View compatible software versions.