BioNeMo - MolMIM Inferencing for Generative Chemistry
Contents
BioNeMo - MolMIM Inferencing for Generative Chemistry#
This tutorial describes the process of runing a Docker container with a pre-trained MolMIM model and using it for generative chemistry. At the end of this tutorial, you will learn:
various functionalities provided in BioNeMo for obtaining chemical embeddings
how to use the learned representations for generative AI and downstream predictive modeling tasks
For a detailed tutorial focusing specifically on property-guided molecular generation with the MolMIM model, please refer to cma_es_guided_molecular_optimization_molmim.ipynb.
Setting up MolMIM#
Prerequisites:#
BioNeMo Framework container is running (refer to the Quickstart Guide)
Familiarity with some components of the BioNeMo framework such as the Models and Inferencing
Assumption: This notebook is being executed from within the BioNeMo docker image.
Downloading pre-trained models#
python download_model.py --download_dir /workspace/bionemo/models/molecule/molmim molmim_70m_24_3
Copy this code and input files into JupyterLab#
In the launched JupyterLab instance, run the Jupyter notebook provided in the code cells below, and copy the benchmark_MoleculeNet_ESOL.csv to the Jupyter-Lab working directory.
Overview#
This walk-through will cover the following aspects of BioNeMo-MolMIM:
BioNeMo-MolMIM Inference Functions:
In this section, we will explore the key inference functionalities of the pre-trained model accessible through the
Inferencewrapper over the model.
These functionalities include obtaining latent space representations with seqs_to_hidden, embeddings with seqs_to_embedding, using input SMILES, as well as obtaining SMILES from latent space representations with hidden_to_seqs.
Molecule Generation / Chemical Space Exploration:
Here, we will explore the chemical space around input query compound and generate related compounds.
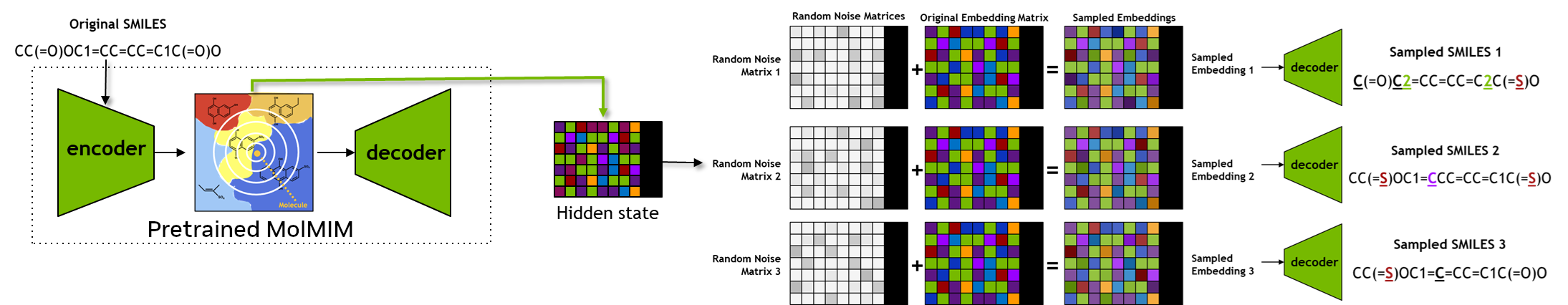
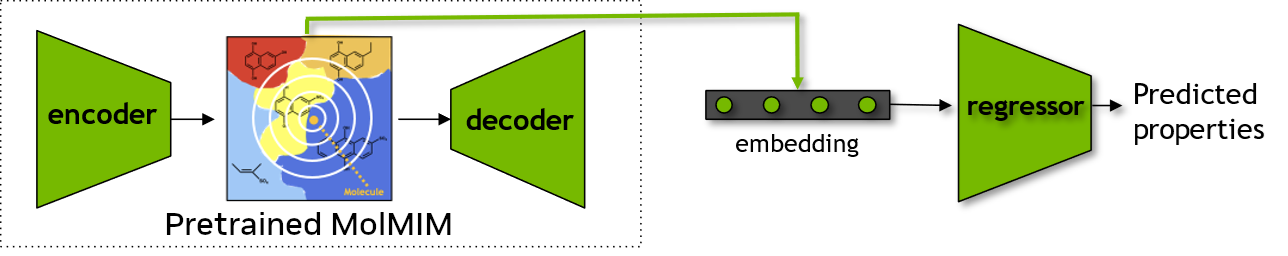
Using Embeddings for Predictive Modeling:
Finally, we will use the embeddings obtained from the BioNeMo-MolMIM for a chemical dataset as features to create a machine learning model to predict properties of the compounds. To evaluate the quality of these embeddings, we will also obtain Morgan Fingerprints to create a comparable baseline prediction model.
Note: The following cells containing python code snippets can be directly copied and executed into a Python environment such as a Jupyter notebook running in a BioNemo container.
Importing required libraries
In the following cell, we import the libraries required to run the tasks for this tutorial.
These imports include open-source CUDA-accelarated data science and machine learning libraries such as cuML, cuDF, cuPY. For more information, check out NVIDIA RAPIDS.
cuDF provides a pandas-like API that will be familiar to data engineers & data scientists, so they can use it to easily accelerate their workflows without going into the details of CUDA programming. Similarly, cuML enables data scientists, researchers, and software engineers to run traditional tabular ML tasks on GPUs. In most cases, cuML’s Python API matches the API from scikit-learn.
Additionally, we will be using RDKit package, which is an open-source cheminformatics library.
# Importing required libraries from RAPIDS, Pandas and NumPy packages
import cuml
import cudf
import cupy as cp
from cuml import LinearRegression
from cuml.svm import SVR
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import pickle
import logging
import warnings
warnings.filterwarnings('ignore')
warnings.simplefilter('ignore')
# Importing libraries from RDKit for handling and manipulating chemical data
from rdkit import Chem
from rdkit.Chem import AllChem, Draw
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import PandasTools
from rdkit.Chem import rdFingerprintGenerator
from rdkit.Chem.Fingerprints import FingerprintMols
from rdkit import RDLogger
RDLogger.DisableLog('rdApp.*')
IPythonConsole.molSize=250,250
Show code cell output
<frozen importlib._bootstrap>:241: RuntimeWarning: pyarrow.lib.IpcWriteOptions size changed, may indicate binary incompatibility. Expected 72 from C header, got 88 from PyObject
<frozen importlib._bootstrap>:241: RuntimeWarning: pyarrow.lib.IpcReadOptions size changed, may indicate binary incompatibility. Expected 96 from C header, got 104 from PyObject
BioNeMo-MolMIM Inference Functions:#
Setup the connection to the inference wrapper and providing example compounds#
In this cell, we create a connection to the MolMIMInference and access the inferencing functions.
MolMIMInference is an adaptor that allows interaction with the inference service of a BioNeMo-MolMIM pre-trained model.
from bionemo.utils.hydra import load_model_config
import os
from bionemo.model.molecule.molmim.infer import MolMIMInference
bionemo_home=f"/workspace/bionemo"
os.environ['BIONEMO_HOME'] = bionemo_home
checkpoint_path = f"{bionemo_home}/models/molecule/molmim/molmim_70m_24_3.nemo"
cfg = load_model_config(config_name="molmim_infer.yaml", config_path=f"{bionemo_home}/examples/tests/conf/") # reasonable starting config for molmim inference
# This is the field of the config that we need to set to our desired checkpoint path.
cfg.model.downstream_task.restore_from_path = checkpoint_path
model = MolMIMInference(cfg, interactive=True)
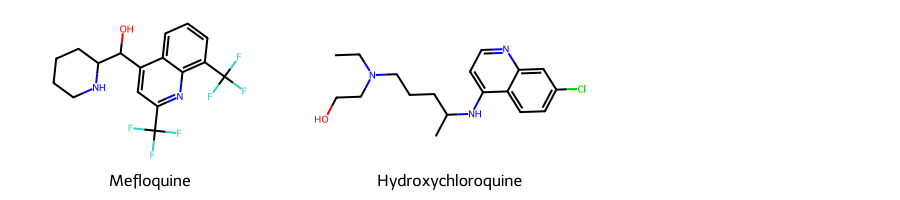
# Here, we are taking two example SMILES for two widely used Antimalarial drugs -- Mefloquine and Hydroxychloroquine
smis = ['OC(c1cc(C(F)(F)F)nc2c(C(F)(F)F)cccc12)C1CCCCN1',
'CCN(CCO)CCCC(C)Nc1ccnc2cc(Cl)ccc12']
# Let us draw these two drugs and see how their 2-D structure looks like, using RD-Kit's functionalities
m1 = Chem.MolFromSmiles(smis[0])
m2 = Chem.MolFromSmiles(smis[1])
Draw.MolsToGridImage((m1,m2), legends=["Mefloquine","Hydroxychloroquine"], subImgSize=(300,200))
Show code cell output
[NeMo I 2024-03-22 16:17:20 megatron_hiddens:110] Registered hidden transform sampled_var_cond_gaussian at bionemo.model.core.hiddens_support.SampledVarGaussianHiddenTransform
[NeMo I 2024-03-22 16:17:20 megatron_hiddens:110] Registered hidden transform interp_var_cond_gaussian at bionemo.model.core.hiddens_support.InterpVarGaussianHiddenTransform
[NeMo I 2024-03-22 16:17:20 utils:326] Restoring model from /workspace/bionemo/models/molecule/molmim/molmim_70m_24_3.nemo
[NeMo I 2024-03-22 16:17:20 utils:330] Loading model class: bionemo.model.molecule.molmim.molmim_model.MolMIMModel
Interactive mode selected, using strategy='auto'
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
HPU available: False, using: 0 HPUs
[NeMo I 2024-03-22 16:17:20 exp_manager:394] Experiments will be logged at /workspace/bionemo/test_results/nemo_experiments/molmim_infer/MolMIM_Inference/2024-03-22_16-17-20
[NeMo I 2024-03-22 16:17:20 utils:299]
************** Trainer configuration ***********
[NeMo I 2024-03-22 16:17:21 utils:300]
name: MolMIM_Inference
desc: Minimum configuration for initializing a MolMIM model for inference.
trainer:
precision: 16-mixed
devices: 1
num_nodes: 1
accelerator: gpu
logger: false
accumulate_grad_batches: 1
exp_manager:
explicit_log_dir: null
exp_dir: ${oc.env:BIONEMO_HOME}/test_results/nemo_experiments/molmim_infer
name: ${name}
create_checkpoint_callback: false
create_wandb_logger: false
create_tensorboard_logger: false
wandb_logger_kwargs:
offline: true
model:
encoder:
num_layers: 6
hidden_size: 512
ffn_hidden_size: 2048
num_attention_heads: 8
init_method_std: 0.02
hidden_dropout: 0.1
attention_dropout: 0.1
ffn_dropout: 0.0
position_embedding_type: learned_absolute
relative_attention_num_buckets: 32
relative_attention_max_distance: 128
relative_position_bias_self_attention_only: true
kv_channels: null
apply_query_key_layer_scaling: false
layernorm_epsilon: 1.0e-05
persist_layer_norm: true
bias_activation_fusion: true
grad_div_ar_fusion: true
masked_softmax_fusion: true
bias_dropout_add_fusion: true
bias: true
normalization: layernorm
arch: perceiver
activation: gelu
headscale: false
transformer_block_type: pre_ln
hidden_steps: 1
num_self_attention_per_cross_attention: 1
openai_gelu: false
onnx_safe: false
fp32_residual_connection: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
activations_checkpoint_granularity: null
megatron_legacy: false
normalize_attention_scores: true
num_moe_experts: 1
moe_frequency: 1
moe_dropout: 0.0
use_flash_attention: false
decoder:
num_layers: 6
hidden_size: 512
ffn_hidden_size: 2048
num_attention_heads: 8
init_method_std: 0.02
hidden_dropout: 0.1
attention_dropout: 0.1
ffn_dropout: 0.0
position_embedding_type: learned_absolute
relative_attention_num_buckets: 32
relative_attention_max_distance: 128
relative_position_bias_self_attention_only: true
kv_channels: null
apply_query_key_layer_scaling: false
layernorm_epsilon: 1.0e-05
persist_layer_norm: true
bias_activation_fusion: true
grad_div_ar_fusion: true
masked_softmax_fusion: true
bias_dropout_add_fusion: true
bias: true
normalization: layernorm
arch: transformer
activation: gelu
headscale: false
transformer_block_type: pre_ln
hidden_steps: 32
num_self_attention_per_cross_attention: 1
openai_gelu: false
onnx_safe: false
fp32_residual_connection: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
activations_checkpoint_granularity: null
megatron_legacy: false
normalize_attention_scores: true
num_moe_experts: 1
moe_frequency: 1
moe_dropout: 0.0
use_flash_attention: false
name: MolMIM-small
micro_batch_size: ${model.data.batch_size}
global_batch_size: 128
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
resume_from_checkpoint: null
pipeline_model_parallel_split_rank: 0
make_vocab_size_divisible_by: 128
pre_process: true
post_process: true
megatron_amp_O2: false
seq_length: 128
max_position_embeddings: 128
gradient_as_bucket_view: true
bias_gelu_fusion: true
share_token_embeddings: true
share_decoder_tokens_head_embeddings: false
hidden_size: 512
training_callbacks: []
hiddens:
enc_output_name: z
enc_inference_output_name: z_mean
token_aggregation_method: mean
hidden_aggregation_method: mean
transform:
q_z_given_x:
cls_name: sampled_var_cond_gaussian
hidden_size: 512
min_logvar: -6.0
max_logvar: 0.0
map_var_to_hiddens: false
loss:
mim:
cls_name: a_mim
loss_weight: 1.0
tokenizer:
library: regex
type: null
model: nemo:048c1f797f464dd5b6a90f60f9405827_molmim.model
vocab_file: nemo:dd344353154640acbbaea1d4536fa7d0_molmim.vocab
merge_file: null
vocab_path: ${oc.env:BIONEMO_HOME}/tokenizers/molecule/molmim/vocab/molmim.vocab
model_path: ${oc.env:BIONEMO_HOME}/tokenizers/molecule/molmim/vocab/molmim.model
data:
links_file: /workspace/bionemo/examples/molecule/megamolbart/dataset/ZINC-downloader.txt
dataset_path: ${oc.env:BIONEMO_HOME}/examples/tests/test_data/molecule/physchem/SAMPL/test/x000
dataset:
train: x_OP_000..175_CL_
test: x_OP_000..175_CL_
val: x_OP_000..004_CL_
canonicalize_target_smile: true
canonicalize_encoder_input: true
canonicalize_decoder_output: true
encoder_augment: false
decoder_independent_augment: false
encoder_mask: false
decoder_mask: false
mask_prob: 0.0
span_lambda: 3.0
micro_batch_size: 2048
num_workers: 4
dataloader_type: single
max_seq_length: 128
seed: 42
skip_lines: 0
drop_last: false
pin_memory: false
data_impl: ''
index_mapping_type: online
data_impl_kwargs:
csv_mmap:
newline_int: 10
header_lines: 1
workers: 10
sort_dataset_paths: true
data_sep: ','
data_col: 1
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
use_upsampling: true
index_mapping_dir: null
batch_size: 128
output_fname: ${oc.env:BIONEMO_HOME}/test_results/nemo_experiments/molmim_infer/x000.pkl
data_fields_map:
sequence: smiles
id: iupac
optim:
name: fused_adam
lr: 0.0005
weight_decay: 0.001
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: 10000.0
constant_steps: 50000.0
max_steps: 1000000
min_lr: 5.0e-05
dwnstr_task_validation:
enabled: false
dataset:
class: bionemo.model.core.dwnstr_task_callbacks.SingleValuePredictionCallback
task_type: regression
infer_target: bionemo.model.molecule.molmim.infer.MolMIMInference
max_seq_length: 128
emb_batch_size: 128
batch_size: 128
num_epochs: 10
shuffle: true
num_workers: 8
dataset_path: /data/physchem/
task_name: SAMPL
dataset:
train: x000
test: x000
sequence_column: smiles
target_column: expt
random_seed: 1234
optim:
name: adam
lr: 0.0001
betas:
- 0.9
- 0.999
eps: 1.0e-08
weight_decay: 0.01
sched:
name: WarmupAnnealing
min_lr: 1.0e-05
last_epoch: -1
warmup_ratio: 0.01
max_steps: 1000
precision: 32
target: bionemo.model.molecule.molmim.molmim_model.MolMIMModel
nemo_version: 1.22.0
downstream_task:
restore_from_path: /workspace/bionemo/models/molecule/molmim/molmim_70m_24_3.nemo
outputs:
- embeddings
target: bionemo.model.molecule.molmim.molmim_model.MolMIMModel
infer_target: bionemo.model.molecule.molmim.infer.MolMIMInference
formatters:
simple:
format: '[%(asctime)s][%(name)s][%(levelname)s] - %(message)s'
handlers:
console:
class: logging.StreamHandler
formatter: simple
stream: ext://sys.stdout
file:
class: logging.FileHandler
formatter: simple
filename: /logs/inference.log
root:
level: INFO
handlers:
- console
disable_existing_loggers: false
infer_config:
name: MolMIM_Inference
desc: Store the infer config in this block so we can pull the model path from it
later.
trainer:
precision: 16-mixed
devices: 1
num_nodes: 1
accelerator: gpu
logger: false
exp_manager:
explicit_log_dir: null
exp_dir: null
name: ${name}
create_checkpoint_callback: false
model:
micro_batch_size: ${model.data.batch_size}
downstream_task:
restore_from_path: ${oc.env:BIONEMO_HOME}/models/molecule/molmim/molmim_70m_24_3.nemo
outputs:
- embeddings
data:
num_workers: 4
batch_size: 128
dataset_path: ${oc.env:BIONEMO_HOME}/examples/tests/test_data/molecule/physchem/SAMPL/test/x000
output_fname: ''
index_mapping_dir: null
data_fields_map:
sequence: smiles
id: iupac
data_impl: ''
data_impl_kwargs:
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
training_callbacks: []
tokenizer:
vocab_path: ${oc.env:BIONEMO_HOME}/tokenizers/molecule/molmim/vocab/molmim.vocab
model_path: ${oc.env:BIONEMO_HOME}/tokenizers/molecule/molmim/vocab/molmim.model
target: bionemo.model.molecule.molmim.molmim_model.MolMIMModel
infer_target: bionemo.model.molecule.molmim.infer.MolMIMInference
formatters:
simple:
format: '[%(asctime)s][%(name)s][%(levelname)s] - %(message)s'
handlers:
console:
class: logging.StreamHandler
formatter: simple
stream: ext://sys.stdout
file:
class: logging.FileHandler
formatter: simple
filename: /logs/inference.log
root:
level: INFO
handlers:
- console
disable_existing_loggers: false
hydra:
searchpath:
- file://${oc.env:BIONEMO_HOME}/examples/conf/
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: use_cpu_initialization in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo I 2024-03-22 16:17:21 megatron_init:234] Rank 0 has data parallel group: [0]
[NeMo I 2024-03-22 16:17:21 megatron_init:237] All data parallel group ranks: [[0]]
[NeMo I 2024-03-22 16:17:21 megatron_init:238] Ranks 0 has data parallel rank: 0
[NeMo I 2024-03-22 16:17:21 megatron_init:246] Rank 0 has model parallel group: [0]
[NeMo I 2024-03-22 16:17:21 megatron_init:247] All model parallel group ranks: [[0]]
[NeMo I 2024-03-22 16:17:21 megatron_init:257] Rank 0 has tensor model parallel group: [0]
[NeMo I 2024-03-22 16:17:21 megatron_init:261] All tensor model parallel group ranks: [[0]]
[NeMo I 2024-03-22 16:17:21 megatron_init:262] Rank 0 has tensor model parallel rank: 0
[NeMo I 2024-03-22 16:17:21 megatron_init:276] Rank 0 has pipeline model parallel group: [0]
[NeMo I 2024-03-22 16:17:21 megatron_init:288] Rank 0 has embedding group: [0]
[NeMo I 2024-03-22 16:17:21 megatron_init:294] All pipeline model parallel group ranks: [[0]]
[NeMo I 2024-03-22 16:17:21 megatron_init:295] Rank 0 has pipeline model parallel rank 0
[NeMo I 2024-03-22 16:17:21 megatron_init:296] All embedding group ranks: [[0]]
[NeMo I 2024-03-22 16:17:21 megatron_init:297] Rank 0 has embedding rank: 0
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: use_cpu_initialization in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 megatron_base_model:821] The model: MolMIMModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-03-22 16:17:21 modelPT:251] You tried to register an artifact under config key=tokenizer.vocab_file but an artifact for it has already been registered.
[NeMo I 2024-03-22 16:17:21 tokenizer_utils:199] Using regex tokenization
[NeMo I 2024-03-22 16:17:21 regex_tokenizer:240] Loading vocabulary from file = /tmp/tmpzuw91mel/dd344353154640acbbaea1d4536fa7d0_molmim.vocab
[NeMo I 2024-03-22 16:17:21 regex_tokenizer:254] Loading regex from file = /tmp/tmpzuw91mel/048c1f797f464dd5b6a90f60f9405827_molmim.model
[NeMo I 2024-03-22 16:17:21 megatron_base_model:315] Padded vocab_size: 640, original vocab_size: 523, dummy tokens: 117.
[NeMo I 2024-03-22 16:17:21 megatron_hiddens:121] NOTE: Adding hiddens transforms and losses
[NeMo I 2024-03-22 16:17:21 megatron_hiddens:149] Added transform q_z_given_x with cfg={'cls_name': 'sampled_var_cond_gaussian', 'hidden_size': 512, 'min_logvar': -6.0, 'max_logvar': 0.0, 'map_var_to_hiddens': False}
[NeMo I 2024-03-22 16:17:21 megatron_hiddens:177] Added loss mim with cfg={'cls_name': 'a_mim', 'loss_weight': 1.0}
[NeMo I 2024-03-22 16:17:21 nlp_overrides:752] Model MolMIMModel was successfully restored from /workspace/bionemo/models/molecule/molmim/molmim_70m_24_3.nemo.
[NeMo I 2024-03-22 16:17:22 megatron_lm_encoder_decoder_model:1195] Decoding using the greedy-search method...
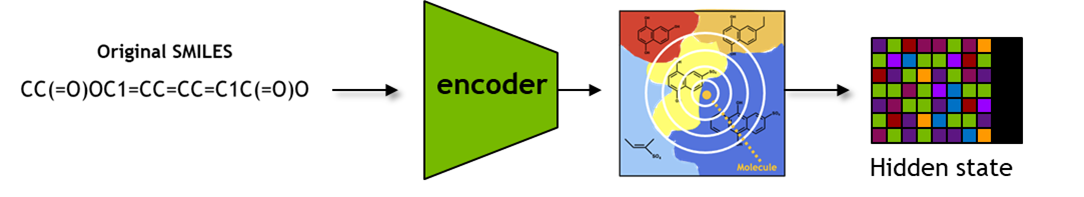
SMILES to Embedding#
Here, we use the seqs_to_embedding function to query the model and fetch the encoder embedding for the input SMILES.
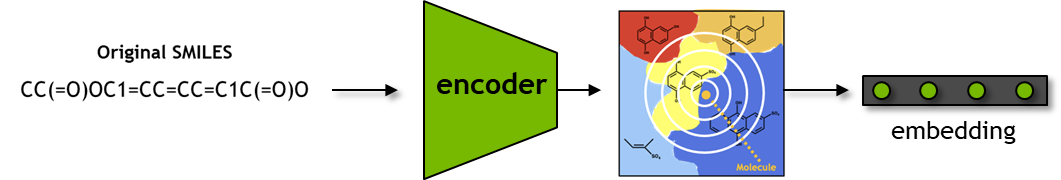
We obtain the learned embeddings for these two compounds below.
embedding = model.seq_to_embeddings(smis)
We will check the shapes of obtained embeddings in the cell below.
embedding.shape
torch.Size([2, 512])
Molecule generation and chemical space exploration#
In this section, we will use the pretrained BioNeMo-MolMIM model to generate designs of novel small-molecules which are similar to the query compound(s).
First, we obtain the hidden state representation for the query compounds using the seqs_to_hiddens functionality, as described in the previous section.
Once the hidden state(s) are obtained, we will use the function chem_sample, as defined below, to manipulate the hidden states and decoding those to generate new chemical designs.
Define chem_sample - the generation function#
The chem_sample function will take a query SMILES as an input, and will return a valid and unique set of generated SMILES.
chem_sample function has two main components.
Obtain the hidden state representation for the input SMILES, perturb copies of this hidden states, and decode those perturbed hidden states to obtain new SMILES.
Using RDKit, check the uniqueness of the generated SMILES set and retain those that are valid SMILES.
# Importing PyTorch library (for more details: https://pytorch.org/)
import torch
# Defining the chemical sampling/generation function
def chem_sample(smis):
# Note, we can control what kind of sampling we want to do using the sampling_kwargs dictionary. Since these are language models, we can use beam-search, top-k, or nucleus sampling.
sampler_kwargs = {
"beam_size": 3, "keep_only_best_tokens": True, "return_scores": False
}
num_samples = 20 # Maximum number of generated molecules per query compound
scaled_radius = 0.7 # Radius of exploration [range: 0.0 - 1.0] --- the extent of perturbation of the original hidden state for sampling
population_samples = model.sample(seqs=smis, num_samples=num_samples, scaled_radius=scaled_radius, sampling_method="beam-search-perturbate", **sampler_kwargs)
assert len(population_samples) == len(smis)
assert len(population_samples[0]) == num_samples
# There are len(smis) different samples, each of which is length num_samples, let's uniquify them and remove invalid ones
uniq_canonical_smiles = []
for smis_samples, original in zip(population_samples, smis):
smis_samples = set(smis_samples) - set([original]) # unique strings that are not the same as we started from
valid_molecules = []
for smis in smis_samples:
mol = Chem.MolFromSmiles(smis)
if mol:
valid_molecules.append(Chem.MolToSmiles(mol,True))
uniq_canonical_smiles.append(valid_molecules)
return uniq_canonical_smiles
Generating analogous small molecules and visualizing them#
In this step, we will use the same two example drug molecules – mefloquine and hydroxychloroquine – for generating new analogues.
# The example SMILES for two widely used Antimalarial drugs -- mefloquine and hydroxychloroquine
smis_lst = ['OC(c1cc(C(F)(F)F)nc2c(C(F)(F)F)cccc12)C1CCCCN1',
'CCN(CCO)CCCC(C)Nc1ccnc2cc(Cl)ccc12']
# Using the chem_sample function and providing smis as input
gen_smis_lst = chem_sample(smis_lst)
for ori_smis, gen_smis in zip(smis_lst, gen_smis_lst):
print(f"Original SMILES: {ori_smis}")
print(f"Generated {len(gen_smis)} unique/valid SMILES: {gen_smis}")
print("\n")
[NeMo I 2024-03-22 16:17:28 megatron_lm_encoder_decoder_model:1192] Decoding using the beam search method with beam size=3...
Original SMILES: OC(c1cc(C(F)(F)F)nc2c(C(F)(F)F)cccc12)C1CCCCN1
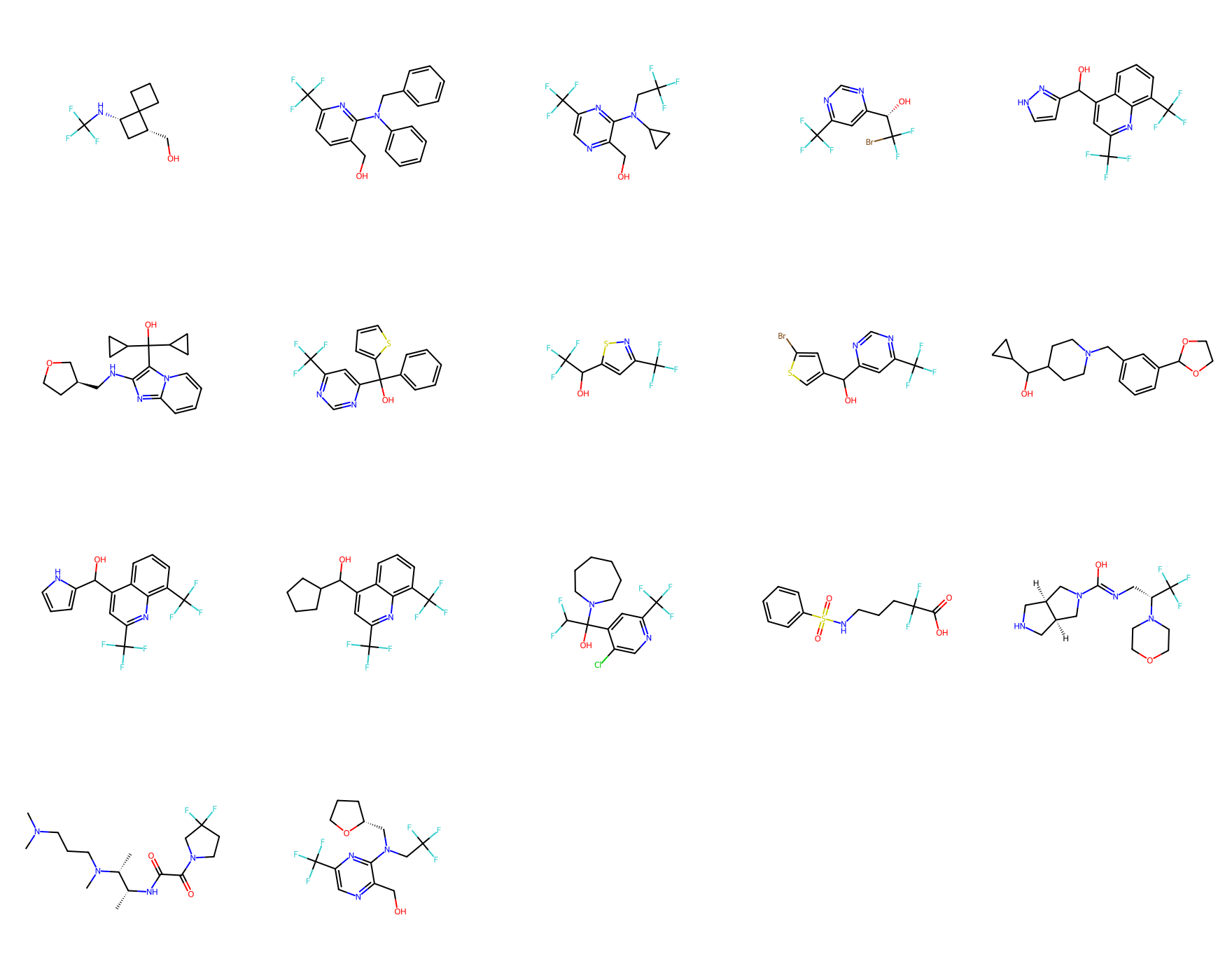
Generated 17 unique/valid SMILES: ['OCc1ncc(C(F)(F)F)nc1N(CC(F)(F)F)C1CC1', 'O[C@@H](c1cc(C(F)(F)F)ncn1)C(F)(F)Br', 'OC(c1c(NC[C@H]2CCOC2)nc2ccccn12)(C1CC1)C1CC1', 'OC(=NC[C@@H](N1CCOCC1)C(F)(F)F)N1C[C@H]2CNC[C@H]2C1', 'OC(c1ccc[nH]1)c1cc(C(F)(F)F)nc2c(C(F)(F)F)cccc12', 'OC(C1CC1)C1CCN(Cc2cccc(C3OCCO3)c2)CC1', 'C[C@H]([C@@H](C)NC(=O)C(=O)N1CCC(F)(F)C1)N(C)CCCN(C)C', 'OC(c1ccccc1)(c1cc(C(F)(F)F)ncn1)c1cccs1', 'OC(c1cc[nH]n1)c1cc(C(F)(F)F)nc2c(C(F)(F)F)cccc12', 'OCc1ncc(C(F)(F)F)nc1N(C[C@H]1CCCO1)CC(F)(F)F', 'OCc1ccc(C(F)(F)F)nc1N(Cc1ccccc1)c1ccccc1', 'O=C(O)C(F)(F)CCCNS(=O)(=O)c1ccccc1', 'OC(c1csc(Br)c1)c1cc(C(F)(F)F)ncn1', 'OC(c1cc(C(F)(F)F)nc2c(C(F)(F)F)cccc12)C1CCCC1', 'OC(c1cc(C(F)(F)F)ncc1Cl)(C(F)F)N1CCCCCC1', 'OC(c1cc(C(F)(F)F)ns1)C(F)(F)F', 'OC[C@@H]1C[C@H](NC(F)(F)F)C12CCC2']
Original SMILES: CCN(CCO)CCCC(C)Nc1ccnc2cc(Cl)ccc12
Generated 14 unique/valid SMILES: ['CCN(CCO)CCNC(=O)N1CCN2c3c(Cl)cccc3C[C@H]2C1', 'CCN(CCO)CCCCN(C)c1ccnc2cc(Cl)ccc12', 'CCN(CCO)CCNC(=O)c1ccnc2cc(Cl)ccc12', 'CCN(CCO)CCCC(=O)Nc1cnc2cc(Cl)ccc2n1', 'CCN(CCO)CCCC(=O)N1CCSc2cc(Cl)ccc21', 'CCN(CCO)CCC(=O)Nc1ccnc2cc(Cl)ccc12', 'CCN(CCO)CCCC(=O)N[C@H]1CCc2cc(Cl)ccc21', 'CCN(CCO)CCCC[S@+]([O-])c1ccnc2cc(Cl)ccc12', 'CCN(CCO)CCCC1CCN(C[C@H](C)c2ccc(F)cc2)CC1', 'CCN(CCO)CCCCNC(=O)c1nc2cc(Cl)ccc2s1', 'CCN(CCO)CCCC(=O)NCc1nc2cc(Cl)ccc2n1C', 'C[C@@H]1CCCC[C@@H]1NCC(=O)N1CCNc2cc(Cl)ccc21', 'CCN(CCO)CCC[C@H](C)Nc1ccnc2cc(Cl)ccc12', 'CCN(CCO)CCC[C@@H](C)Nc1ccnc2cc(Cl)ccc12']
Now, we will take a look at the generated designs.
First lets visualize the generated designs analogous to the first query (mefloquine).
mols_from_gen_smis = [Chem.MolFromSmiles(smi) for smi in set(gen_smis_lst[0])]
print("Total unique molecule designs obtained: ", len(mols_from_gen_smis))
Draw.MolsToGridImage(mols_from_gen_smis, molsPerRow=5, subImgSize=(350,350))
Total unique molecule designs obtained: 17
And now from the second molecule (hydroxychloroquine).
mols_from_gen_smis = [Chem.MolFromSmiles(smi) for smi in set(gen_smis_lst[1])]
print("Total unique molecule designs obtained: ", len(mols_from_gen_smis))
Draw.MolsToGridImage(mols_from_gen_smis, molsPerRow=5, subImgSize=(350,350))
Total unique molecule designs obtained: 14
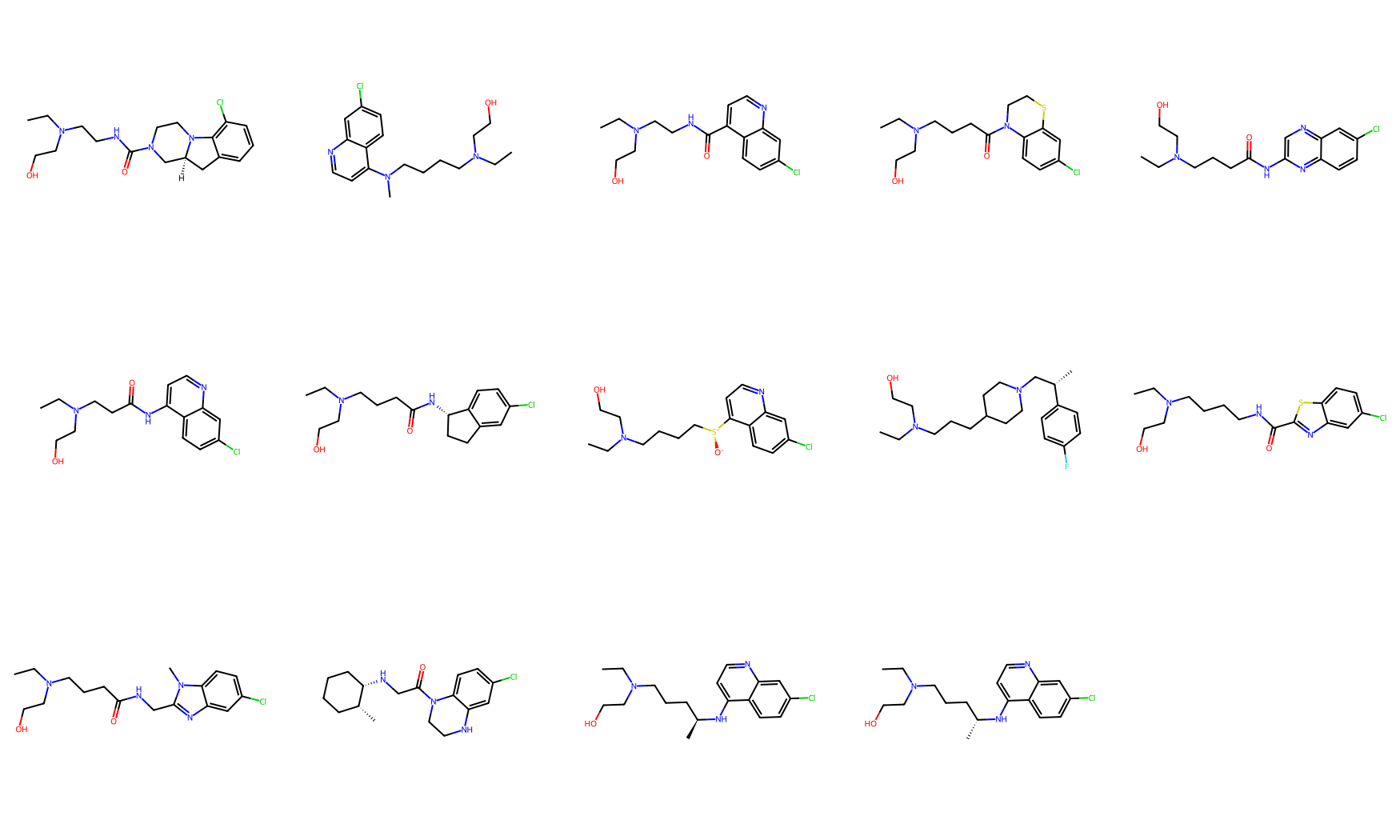
How many molecular designs were obtained from this step?
What would change in the process if you try different values for number of samples and sampling radius in the chem_sample function?
This concludes the Second objective of using the BioNeMo-MolMIM pre-trained model for chemical space exploration and generative chemistry.
Downstream Prediction Model using Learned Embeddings from Pre-trained Model#
One of the improtant tasks for chemoinformaticians is to develop models to predict properties of small molecules.
These properties may include physicochemical parameters, such as lipophilicity, solubility, hydration free energy (LogP, LogD, and so on). These properties can also include certain pharmacokinetic/dynamic behaviors such as Blood-Brain-Barrier/CNS permeability, Vd, etc.
Modeling such properties relies on the selection of appropriate and relevant descriptors/features. In this section, we will use the embeddings from MolMIM as a feature set for training a machine learning model for physico-chemical parameter predictions. We will then compare how this model performs when compared to a model developed using chemical fingerprints (here, Morgan Fingerprints).
In the section below, we use the ESOL dataset curated by MoleculeNet (https://moleculenet.org/datasets-1)
ESOL: Water solubility data(log solubility in mols per litre) for common organic small molecules [n=1129]
Example: Compound Water Solubility (ESOL) Prediction#
In this example, we will use the ESOL dataset from Moleculenet. The dataset is modified for this example purposes to include the relevant columns and placed in /data directory as benchmark_MoleculeNet_ESOL.csv.
We will load the data from benchmark_MoleculeNet_ESOL.csv file in a cuDF dataframe format. After loading the data, we will obtain the embeddings for compounds that are present in the cuDF dataframe by using the previously introduced seqs_to_embedding function of the BioNeMo-MolMIM pretrained model.
Similarly, we will obtain Morgan Fingerprints for the compounds in the dataframe using RDKit’s AllChem.GetMorganFingerprintAsBitVect function.
Finally, we will generate two Support Vector Regression models – using the embeddings and the Morgan Fingerprints – with functions from the cuML library. At the end, we will compare the performances of both the models.
Preprocessing dataset using cuDF#
# Reading the benchmark_MoleculeNet_ESOL.csv file as cuDF DataFrame format
ex_data_file = './benchmark_MoleculeNet_ESOL.csv'
ex_df = cudf.read_csv(ex_data_file)
# Checking the dimensions of the dataframe and the first few rows
print(ex_df.shape)
ex_df.head()
(1128, 4)
| index | task | SMILES | measured log solubility in mols per litre | |
|---|---|---|---|---|
| 0 | 0 | ESOL | Cc1cccc(C)c1O | -1.290 |
| 1 | 1 | ESOL | ClCC(Cl)(Cl)Cl | -2.180 |
| 2 | 2 | ESOL | CC34CCC1C(=CCc2cc(O)ccc12)C3CCC4=O | -5.282 |
| 3 | 3 | ESOL | c1ccc2c(c1)ccc3c2ccc4c5ccccc5ccc43 | -7.870 |
| 4 | 4 | ESOL | CCCCCCCC(=O)C | -2.580 |
Obtaining the embeddings using seqs_to_embedding#
%%time
# Generating embeddings for the compounds in the dataframe.
# We process batches of 100 compounds at a time.
start = 0
ex_emb_df = cudf.DataFrame()
while start < ex_df.shape[0]:
smis = ex_df.iloc[start: start+100, 2]
x_val = ex_df.iloc[start: start+100, 3]
embedding = model.seq_to_embeddings(smis.to_arrow().to_pylist())
ex_emb_df = cudf.concat([ex_emb_df,
cudf.DataFrame({"SMILES": smis,
"EMBEDDINGS": embedding.tolist(),
"Y": x_val})]) # The ESOL value is captured in the 'Y' column
start = start + 100
CPU times: user 321 ms, sys: 13.4 ms, total: 335 ms
Wall time: 325 ms
We can now examine at the new column added with the embedding vectors.
ex_emb_df
| SMILES | EMBEDDINGS | Y | |
|---|---|---|---|
| 0 | Cc1cccc(C)c1O | [0.025419697165489197, 0.3563258945941925, -0.... | -1.290 |
| 1 | ClCC(Cl)(Cl)Cl | [0.3793778419494629, -0.0816623866558075, 0.50... | -2.180 |
| 2 | CC34CCC1C(=CCc2cc(O)ccc12)C3CCC4=O | [-0.9475264549255371, -0.1938450038433075, -0.... | -5.282 |
| 3 | c1ccc2c(c1)ccc3c2ccc4c5ccccc5ccc43 | [-0.2649092674255371, -0.1303684413433075, 0.1... | -7.870 |
| 4 | CCCCCCCC(=O)C | [0.1050858348608017, 0.3077419102191925, 0.017... | -2.580 |
| ... | ... | ... | ... |
| 1123 | OCCCC=C | [0.2729325294494629, 0.2718532383441925, 0.049... | -0.150 |
| 1124 | Clc1ccccc1I | [-0.2469649463891983, -0.0941135585308075, -0.... | -3.540 |
| 1125 | CCC(CC)C=O | [0.0904373973608017, -0.4945041835308075, 0.35... | -1.520 |
| 1126 | Clc1ccc(Cl)cc1 | [0.1441483348608017, -0.3509494960308075, -0.0... | -3.270 |
| 1127 | c1ccc2c(c1)ccc3c4ccccc4ccc23 | [-0.2494063526391983, -0.1621067225933075, -0.... | -8.057 |
1128 rows × 3 columns
Obtaining the Morgan Fingerprints using RDKit functionalities#
%%time
# Here, we will define a function to return Morgan Fingerprints for a list of input SMILES
def get_fp_arr(df_smi):
fp_array = []
for smi in df_smi:
m = Chem.MolFromSmiles(smi) # Converting SMILES to RD-Kit's MOL format
fp = AllChem.GetMorganFingerprintAsBitVect(m, 3, 1024) # Obtain Morgan Fingerprints as a Bit-vector
fp = np.fromstring(fp.ToBitString(), 'u1') - ord('0') # Converting Bit-vector to string type
fp_array.append(fp)
fp_array = np.asarray(fp_array)
return fp_array.tolist()
# Passing the SMILES list to the get_fp_arr function, and saving the returned list of Morgan Fingerprints as a DataFrame column named 'FINGERPRINTS'
ex_emb_df['FINGERPRINT'] = get_fp_arr(ex_emb_df['SMILES'].to_arrow().to_pylist())
# Let's take a look at the DataFrame after Morgan_Fingerprint calculation
ex_emb_df
CPU times: user 164 ms, sys: 31.3 ms, total: 195 ms
Wall time: 182 ms
| SMILES | EMBEDDINGS | Y | FINGERPRINT | |
|---|---|---|---|---|
| 0 | Cc1cccc(C)c1O | [0.025419697165489197, 0.3563258945941925, -0.... | -1.290 | [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| 1 | ClCC(Cl)(Cl)Cl | [0.3793778419494629, -0.0816623866558075, 0.50... | -2.180 | [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| 2 | CC34CCC1C(=CCc2cc(O)ccc12)C3CCC4=O | [-0.9475264549255371, -0.1938450038433075, -0.... | -5.282 | [0, 0, 0, 1, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, ... |
| 3 | c1ccc2c(c1)ccc3c2ccc4c5ccccc5ccc43 | [-0.2649092674255371, -0.1303684413433075, 0.1... | -7.870 | [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| 4 | CCCCCCCC(=O)C | [0.1050858348608017, 0.3077419102191925, 0.017... | -2.580 | [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| ... | ... | ... | ... | ... |
| 1123 | OCCCC=C | [0.2729325294494629, 0.2718532383441925, 0.049... | -0.150 | [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| 1124 | Clc1ccccc1I | [-0.2469649463891983, -0.0941135585308075, -0.... | -3.540 | [0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| 1125 | CCC(CC)C=O | [0.0904373973608017, -0.4945041835308075, 0.35... | -1.520 | [0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| 1126 | Clc1ccc(Cl)cc1 | [0.1441483348608017, -0.3509494960308075, -0.0... | -3.270 | [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
| 1127 | c1ccc2c(c1)ccc3c4ccccc4ccc23 | [-0.2494063526391983, -0.1621067225933075, -0.... | -8.057 | [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ... |
1128 rows × 4 columns
Generating ML models using cuML#
Now that we have both the embeddings and the Morgan Fingerprints calculated for the ESOL dataset, we can use the cuML to generate a prediction model.
Here, we will be creating a Support Vector Regression model. For more details, refer to https://docs.rapids.ai/api/cuml/stable/api.html#support-vector-machines.
First, let’s make a model using Morgan Fingerprints:
### Using Morgan fingerprints for developing a Support Vector Regression prediction model ###
# Splitting the input dataset into Training (70%) and Test sets(30%)
tempX = cp.asarray(ex_emb_df['FINGERPRINT'].to_arrow().to_pylist(), dtype=cp.float32)
tempY = cp.asarray(ex_emb_df['Y'], dtype=cp.float32)
x_train, x_test_mfp, y_train, y_test_mfp = cuml.train_test_split(tempX, tempY, train_size=0.7, random_state=1993)
# Defining Support vector regression model parameters
reg_mfp = SVR(kernel='rbf', gamma='scale', C=10, epsilon=0.1) # You may change the model parameters and observe the change in the performance
# Fitting the model on the training dataset
reg_mfp.fit(x_train, y_train)
SVR()
# Using the fitted model for prediction of the ESOL values for the test dataset compounds
pred_mfp = reg_mfp.predict(x_test_mfp)
# Performance measures of SVR model - Mean Squared Error and R-squared values
mfp_SVR_MSE = cuml.metrics.mean_squared_error(y_test_mfp, pred_mfp)
mfp_SVR_R2 = cuml.metrics.r2_score(y_test_mfp, pred_mfp)
print("Fingerprint_SVR_MSE: ", mfp_SVR_MSE)
print("Fingerprint_SVR_r2: ", mfp_SVR_R2)
Fingerprint_SVR_MSE: 1.1670269
Fingerprint_SVR_r2: 0.740040123462677
Now, we can use BioNeMo-MolMIM Embeddings to develop a Support Vector Regression prediction model.
### Using BioNeMo-MolMIM Embeddings for developing a SVR model ###
# Splitting dataset into training and testing sets
tempX = cp.asarray(ex_emb_df['EMBEDDINGS'].to_arrow().to_pylist(), dtype=cp.float32)
tempY = cp.asarray(ex_emb_df['Y'], dtype=cp.float32)
x_train, x_test_emb, y_train, y_test_emb = cuml.train_test_split(tempX, tempY, train_size=0.7, random_state=1993)
# Defining Support vector regression model parameters
reg_emb = SVR(kernel='rbf', gamma='scale', C=10, epsilon=0.1) # You may change the model parameters and observe the change in the performance
# Fitting the model on the training dataset
reg_emb.fit(x_train, y_train)
# Using the fitted model for prediction of the ESOL values for the test dataset compounds
pred_emb = reg_emb.predict(x_test_emb)
# Performance measures of SVR model
emb_SVR_MSE = cuml.metrics.mean_squared_error(y_test_emb, pred_emb)
emb_SVR_R2 = cuml.metrics.r2_score(y_test_emb, pred_emb)
print("Embeddings_SVR_MSE: ", emb_SVR_MSE)
print("Embeddings_SVR_r2: ", emb_SVR_R2)
Embeddings_SVR_MSE: 1.2011706
Embeddings_SVR_r2: 0.7324345111846924
Let’s plot the MSE and R-squared values obtained for both the models.
import matplotlib.pyplot as plt
plt.rcParams.update({'font.size': 14})
data = [[cp.asnumpy(mfp_SVR_MSE), mfp_SVR_R2],
[cp.asnumpy(emb_SVR_MSE), emb_SVR_R2]]
X = np.arange(2)
fig = plt.figure()
ax = fig.add_axes([0,0,1,1])
ax.bar(X + 0.00, data[0], color = 'b', width = 0.20)
ax.bar(X + 0.20, data[1], color = '#76b900', width = 0.20)
ax.set_xticks([0.1, 1.1])
ax.set_xticklabels(['[MSE] \n Mean Squared Error', '$R^2$'])
ax.legend(['Morgan Fingerprints', 'MolMIM Embeddings'],loc='best')
<matplotlib.legend.Legend at 0x7fd07f11b670>
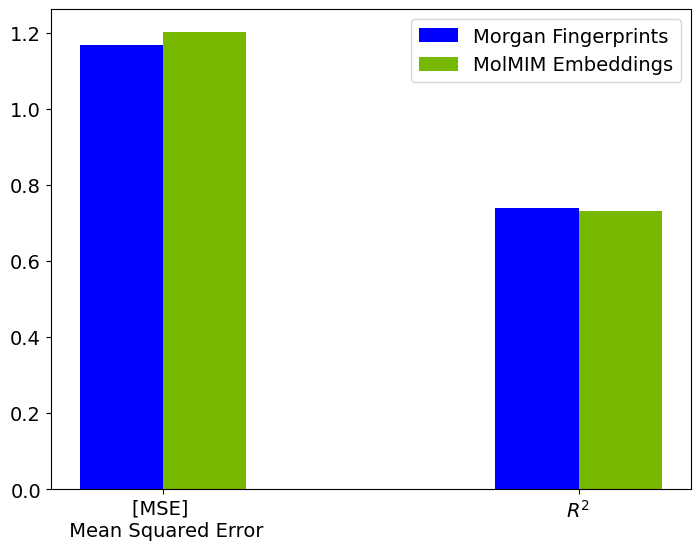
We can also examine the experimental and predicted ESOL values from both regressors trained on different featurizations.
plt.rcParams.update({'font.size': 14})
fig = plt.figure()
ax = fig.add_axes([0,0,1,1])
ax.scatter(cp.asnumpy(y_test_mfp), cp.asnumpy(pred_mfp), s=15, label='Morgan Fingerprints')
ax.scatter(cp.asnumpy(y_test_mfp), cp.asnumpy(pred_emb), s=15, label='MolMIM Embeddings')
ax.grid(alpha=0.3)
ax.plot([-12, 2], [-12, 2], 'r--') # The "perfect prediction" line
ax.set_xlabel('ESOL, experiment')
ax.set_ylabel('ESOL, predicted')
ax.legend()
<matplotlib.legend.Legend at 0x7fd078982050>
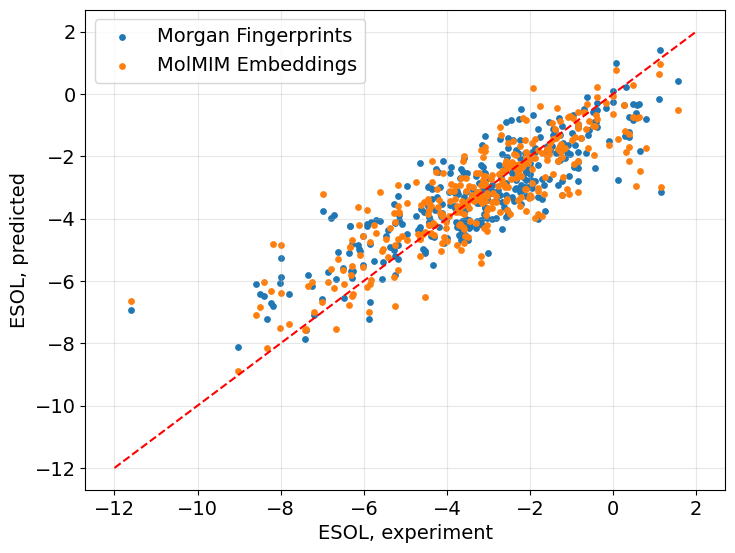
The overall performance of predictions made using the model trained with MolMIM embeddings is comparable to those predictions made with the Morgan Fingerprint-based model.
This concludes the final objective of using the embeddings for predictive modeling of downstream tasks.