BioNeMo - Geneformer inferencing for single cell downstream tasks
Contents
BioNeMo - Geneformer inferencing for single cell downstream tasks#
This tutorial showcases how to run the BioNeMo container, pre-train a geneformer model, and use it for inferencing downstream single cell tasks. At the end of this tutorial, a user will learn:
launching the BioNeMo container
Download data from czi to use for inference.
Convert AnnData files into the sparse CSR memmap format used by BioNeMo
Download a pretrained checkpoint
Restore the geneformer checkpoint and perform inference with the czi dataset.
Prerequisites:#
BioNeMo Framework container is running (refer to the Quickstart Guide)
Familiarity with some components of the BioNeMo framework such as the Models and Inferencing
Running the BioNeMo container#
This example has been built by launching the container in a local machine with 2 x A6000 RTX GPUs. Refer to specific instructions for [remote and multi-node launch]
Once the container is launched, navigate to http://0.0.0.0:8888, http://localhost:8888, or the IP address of the workstation/node. A JupyterLab instance should show up.
Copy this code and input files into JupyterLab#
In the launched JupyterLab, run the codes in a Jupyter notebook as provided in the code cells below.
Getting example single cell data and setting it up for inference#
First, we must acquire single cell training data for inference. To do this we will install the cellxgene-census api and download a small dataset. We use the example provided by the czi api examples page to download a single h5ad file. Generally, our workflow expects a collection of h5ad files to be used for pre-training. In this case, we restrict to 100k cells from a single dataset to keep training time and downloading time small.
!pip install cellxgene-census
Defaulting to user installation because normal site-packages is not writeable
Looking in indexes: https://pypi.org/simple, https://pypi.ngc.nvidia.com
Requirement already satisfied: cellxgene-census in /workspace/bionemo/.local/lib/python3.10/site-packages (1.13.0)
Requirement already satisfied: tiledbsoma~=1.9.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from cellxgene-census) (1.9.5)
Requirement already satisfied: anndata in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (0.10.6)
Requirement already satisfied: numpy>=1.21 in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (1.24.4)
Requirement already satisfied: requests in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (2.31.0)
Requirement already satisfied: typing-extensions in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (4.7.1)
Requirement already satisfied: s3fs>=2021.06.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from cellxgene-census) (2024.3.1)
Requirement already satisfied: aiobotocore<3.0.0,>=2.5.4 in /workspace/bionemo/.local/lib/python3.10/site-packages (from s3fs>=2021.06.1->cellxgene-census) (2.12.3)
Requirement already satisfied: fsspec==2024.3.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from s3fs>=2021.06.1->cellxgene-census) (2024.3.1)
Requirement already satisfied: aiohttp!=4.0.0a0,!=4.0.0a1 in /usr/local/lib/python3.10/dist-packages (from s3fs>=2021.06.1->cellxgene-census) (3.9.0)
Requirement already satisfied: attrs>=22.2 in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (23.1.0)
Requirement already satisfied: numba>=0.58.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from tiledbsoma~=1.9.1->cellxgene-census) (0.59.1)
Requirement already satisfied: pandas in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.5.3)
Requirement already satisfied: pyarrow-hotfix in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (0.6)
Requirement already satisfied: scanpy>=1.9.2 in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.9.8)
Requirement already satisfied: scipy in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.11.1)
Requirement already satisfied: somacore==1.0.10 in /workspace/bionemo/.local/lib/python3.10/site-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.0.10)
Requirement already satisfied: tiledb~=0.27.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from tiledbsoma~=1.9.1->cellxgene-census) (0.27.1)
Requirement already satisfied: pyarrow>=9.0.0 in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (14.0.1)
Requirement already satisfied: array-api-compat!=1.5,>1.4 in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (1.5.1)
Requirement already satisfied: exceptiongroup in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (1.1.3)
Requirement already satisfied: h5py>=3.1 in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (3.10.0)
Requirement already satisfied: natsort in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (8.4.0)
Requirement already satisfied: packaging>=20.0 in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (23.1)
Requirement already satisfied: charset-normalizer<4,>=2 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (3.2.0)
Requirement already satisfied: idna<4,>=2.5 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (3.4)
Requirement already satisfied: urllib3<3,>=1.21.1 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (1.26.16)
Requirement already satisfied: certifi>=2017.4.17 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (2023.7.22)
Requirement already satisfied: botocore<1.34.70,>=1.34.41 in /workspace/bionemo/.local/lib/python3.10/site-packages (from aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (1.34.69)
Requirement already satisfied: wrapt<2.0.0,>=1.10.10 in /usr/local/lib/python3.10/dist-packages (from aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (1.14.1)
Requirement already satisfied: aioitertools<1.0.0,>=0.5.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (0.11.0)
Requirement already satisfied: multidict<7.0,>=4.5 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (6.0.4)
Requirement already satisfied: yarl<2.0,>=1.0 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (1.9.2)
Requirement already satisfied: frozenlist>=1.1.1 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (1.4.0)
Requirement already satisfied: aiosignal>=1.1.2 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (1.3.1)
Requirement already satisfied: async-timeout<5.0,>=4.0 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (4.0.3)
Requirement already satisfied: llvmlite<0.43,>=0.42.0dev0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from numba>=0.58.0->tiledbsoma~=1.9.1->cellxgene-census) (0.42.0)
Requirement already satisfied: python-dateutil>=2.8.1 in /usr/local/lib/python3.10/dist-packages (from pandas->tiledbsoma~=1.9.1->cellxgene-census) (2.8.2)
Requirement already satisfied: pytz>=2020.1 in /usr/local/lib/python3.10/dist-packages (from pandas->tiledbsoma~=1.9.1->cellxgene-census) (2023.3)
Requirement already satisfied: joblib in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.2.0)
Requirement already satisfied: matplotlib>=3.6 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (3.8.0)
Requirement already satisfied: networkx>=2.3 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (2.6.3)
Requirement already satisfied: patsy in /workspace/bionemo/.local/lib/python3.10/site-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.5.6)
Requirement already satisfied: scikit-learn>=0.24 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.2.0)
Requirement already satisfied: seaborn>=0.13.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.13.2)
Requirement already satisfied: session-info in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.0.0)
Requirement already satisfied: statsmodels>=0.10.0rc2 in /workspace/bionemo/.local/lib/python3.10/site-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.14.1)
Requirement already satisfied: tqdm in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (4.66.1)
Requirement already satisfied: umap-learn>=0.3.10 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.5.5)
Requirement already satisfied: jmespath<2.0.0,>=0.7.1 in /usr/local/lib/python3.10/dist-packages (from botocore<1.34.70,>=1.34.41->aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (1.0.1)
Requirement already satisfied: contourpy>=1.0.1 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.1.1)
Requirement already satisfied: cycler>=0.10 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.12.1)
Requirement already satisfied: fonttools>=4.22.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (4.51.0)
Requirement already satisfied: kiwisolver>=1.0.1 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.4.5)
Requirement already satisfied: pillow>=6.2.0 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (10.2.0)
Requirement already satisfied: pyparsing>=2.3.1 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (3.1.1)
Requirement already satisfied: six>=1.5 in /usr/local/lib/python3.10/dist-packages (from python-dateutil>=2.8.1->pandas->tiledbsoma~=1.9.1->cellxgene-census) (1.16.0)
Requirement already satisfied: threadpoolctl>=2.0.0 in /usr/local/lib/python3.10/dist-packages (from scikit-learn>=0.24->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (3.2.0)
Requirement already satisfied: pynndescent>=0.5 in /usr/local/lib/python3.10/dist-packages (from umap-learn>=0.3.10->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.5.11)
Requirement already satisfied: stdlib-list in /usr/local/lib/python3.10/dist-packages (from session-info->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.10.0)
[notice] A new release of pip is available: 23.2.1 -> 24.0
[notice] To update, run: python -m pip install --upgrade pip
# Below are paths required for setting up pre-training and inference.
tutorial_data_dir = "/workspace/bionemo/data/singlecell_inf_tutorial/download_anndata"
tutorial_processed_dir = "/workspace/bionemo/data/singlecell_inf_tutorial/processed_data"
tutorial_output_dir = "/workspace/bionemo/data/singlecell_inf_tutorial/inference_output"
tutorial_output_inference_pickle = f"{tutorial_output_dir}/human_covid19_bcells_pretrained_inference.pkl"
demo_data_download_path = f"{tutorial_data_dir}/human_covid19_bcells.h5ad"
!mkdir -p {tutorial_data_dir}
!mkdir -p {tutorial_processed_dir}
!mkdir -p {tutorial_output_dir}
!rm -f {tutorial_output_inference_pickle} # clean this up if it's already there
!rm -rf {tutorial_processed_dir}
import cellxgene_census
with cellxgene_census.open_soma(census_version="2023-12-15") as census:
filter1 = "cell_type == 'B cell' and tissue_general == 'lung' and disease == 'COVID-19' and is_primary_data == True"
adata = cellxgene_census.get_anndata(
census = census,
organism = "Homo sapiens",
obs_value_filter = filter1,
)
adata[:100000].write(demo_data_download_path)
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
/usr/local/lib/python3.10/dist-packages/anndata/_core/anndata.py:1209: ImplicitModificationWarning: Trying to modify attribute `.obs` of view, initializing view as actual.
df[key] = c
!ls -laht {demo_data_download_path}
-rw-r--r-- 1 jstjohn domain-users 27M May 13 16:52 /workspace/bionemo/data/singlecell_inf_tutorial/download_anndata/human_covid19_bcells.h5ad
!python /workspace/bionemo/bionemo/data/singlecell/sc_memmap.py \
--data-path {tutorial_data_dir} \
--save-path {tutorial_processed_dir}
Found 1 files
Starting to create memmap files...
Creating metadata...: 100%|███████████████████████| 1/1 [00:00<00:00, 8.48it/s]
Done creating `metadata.json`
Writing data into memmaps to /workspace/bionemo/data/singlecell_inf_tutorial/processed_data...
Merging AnnData into numpy memaps...: 100%|███████| 1/1 [00:00<00:00, 7.31it/s]
Saving dataframe ...
Done creating dataset ...
!ls -laht {tutorial_processed_dir}
total 19M
-rw-r--r-- 1 jstjohn domain-users 194K May 13 16:52 features.csv
drwxr-xr-x 2 jstjohn domain-users 4.0K May 13 16:52 .
-rw-r--r-- 1 jstjohn domain-users 8.5M May 13 16:52 gene_expression_ind.npy
-rw-r--r-- 1 jstjohn domain-users 19K May 13 16:52 gene_expression_ptr.npy
-rw-r--r-- 1 jstjohn domain-users 8.5M May 13 16:52 gene_expression_data.npy
-rw-r--r-- 1 jstjohn domain-users 1.1M May 13 16:52 metadata.json
drwxr-xr-x 5 jstjohn domain-users 4.0K May 13 16:52 ..
Running inference.#
Now we will use a pretrained nemo file and start there for inference. For this tutorial we’ll use the large variant of our pretrained checkpoint, the 103M parameter variant based on BERT-base with enhancements from the geneformer publications.
!cd /workspace/bionemo/ && python download_models.py --download_dir models geneformer_106M_240530 #--source pbss
Running command: ngc --version
NGC CLI 3.38.0
Done.
Warning: geneformer_106M_240530 does not have a ngc URL; skipping download.
pretrained_nemo_file = '/workspace/bionemo/models/singlecell/geneformer/geneformer-106M-240530.nemo'
!python /workspace/bionemo/bionemo/model/infer.py \
--config-dir /workspace/bionemo/examples/singlecell/geneformer/conf \
--config-name infer \
++model.downstream_task.restore_from_path={pretrained_nemo_file} \
++model.data.batch_size=8 \
++model.data.dataset_path={tutorial_processed_dir} \
++exp_manager.exp_dir={tutorial_output_dir} \
++model.data.output_fname={tutorial_output_inference_pickle}
[NeMo W 2024-05-13 16:52:39 nemo_logging:349] /usr/lib/python3.10/multiprocessing/popen_fork.py:66: RuntimeWarning: os.fork() was called. os.fork() is incompatible with multithreaded code, and JAX is multithreaded, so this will likely lead to a deadlock.
self.pid = os.fork()
[NeMo I 2024-05-13 16:52:40 megatron_hiddens:110] Registered hidden transform sampled_var_cond_gaussian at bionemo.model.core.hiddens_support.SampledVarGaussianHiddenTransform
[NeMo I 2024-05-13 16:52:40 megatron_hiddens:110] Registered hidden transform interp_var_cond_gaussian at bionemo.model.core.hiddens_support.InterpVarGaussianHiddenTransform
[NeMo W 2024-05-13 16:52:41 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/hydra/_internal/hydra.py:119: UserWarning: Future Hydra versions will no longer change working directory at job runtime by default.
See https://hydra.cc/docs/1.2/upgrades/1.1_to_1.2/changes_to_job_working_dir/ for more information.
ret = run_job(
[NeMo I 2024-05-13 16:52:41 loading:31]
************** Experiment configuration ***********
[NeMo I 2024-05-13 16:52:41 loading:32]
name: geneformer_inference
desc: Minimum configuration for initializing a Geneformer model for inference.
trainer:
precision: bf16-mixed
devices: 1
num_nodes: 1
accelerator: gpu
logger: false
exp_manager:
explicit_log_dir: null
exp_dir: /workspace/bionemo/data/singlecell_inf_tutorial/inference_output
name: ${name}
create_checkpoint_callback: false
model:
micro_batch_size: ${model.data.batch_size}
downstream_task:
restore_from_path: /workspace/bionemo/models/singlecell/geneformer/geneformer-106M-240530.nemo
outputs:
- embeddings
- hiddens
data:
num_workers: 4
batch_size: 8
dataset_path: /workspace/bionemo/data/singlecell_inf_tutorial/processed_data
output_fname: /workspace/bionemo/data/singlecell_inf_tutorial/inference_output/human_covid19_bcells_pretrained_inference.pkl
index_mapping_dir: null
data_fields_map:
sequence: sequence
id: id
data_impl: geneformer
data_impl_kwargs:
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
dynamic_padding: true
post_process: false
inference_output_everything: false
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
infer_target: bionemo.model.singlecell.geneformer.infer.GeneformerInference
formatters:
simple:
format: '[%(asctime)s][%(name)s][%(levelname)s] - %(message)s'
handlers:
console:
class: logging.StreamHandler
formatter: simple
stream: ext://sys.stdout
file:
class: logging.FileHandler
formatter: simple
filename: /logs/inference.log
root:
level: INFO
handlers:
- console
disable_existing_loggers: false
[NeMo I 2024-05-13 16:52:41 utils:333] Restoring model from /workspace/bionemo/models/singlecell/geneformer/geneformer-106M-240530.nemo
[NeMo I 2024-05-13 16:52:41 utils:337] Loading model class: bionemo.model.singlecell.geneformer.model.GeneformerModel
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
HPU available: False, using: 0 HPUs
[NeMo I 2024-05-13 16:52:41 exp_manager:394] Experiments will be logged at /workspace/bionemo/data/singlecell_inf_tutorial/inference_output/geneformer_inference/2024-05-13_16-52-41
[NeMo I 2024-05-13 16:52:41 exp_manager:835] TensorboardLogger has been set up
[NeMo I 2024-05-13 16:52:41 utils:306]
************** Trainer configuration ***********
[NeMo I 2024-05-13 16:52:41 utils:307]
name: geneformer_inference
desc: Minimum configuration for initializing a Geneformer model for inference.
trainer:
precision: bf16-mixed
devices: 1
num_nodes: 1
accelerator: gpu
logger: false
accumulate_grad_batches: 1
exp_manager:
explicit_log_dir: null
exp_dir: /workspace/bionemo/data/singlecell_inf_tutorial/inference_output
name: ${name}
create_checkpoint_callback: false
model:
tokenizer:
vocab_file: nemo:e0c840a303ab4ce7a8ad36400266741d_geneformer.vocab
micro_batch_size: ${model.data.batch_size}
activation: relu
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
use_flash_attention: true
seq_length: 2048
encoder_seq_length: 2048
max_position_embeddings: 2048
num_layers: 12
hidden_size: 768
ffn_hidden_size: 3072
num_attention_heads: 12
init_method_std: 0.02
hidden_dropout: 0.02
attention_dropout: 0.02
kv_channels: null
apply_query_key_layer_scaling: true
layernorm_epsilon: 1.0e-12
make_vocab_size_divisible_by: 128
pre_process: true
post_process: false
bert_binary_head: false
resume_from_checkpoint: null
masked_softmax_fusion: true
native_amp_init_scale: 4294967296
native_amp_growth_interval: 1000
fp32_residual_connection: true
fp16_lm_cross_entropy: false
seed: 1234
use_cpu_initialization: false
onnx_safe: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
data:
data_impl: geneformer
probabilistic_dirichlet_sampling_train: false
train_dataset_path: /workspace/bionemo/data/cellxgene_2023-12-15/processed_data/train
val_dataset_path: /workspace/bionemo/data/cellxgene_2023-12-15/processed_data/val
test_dataset_path: /workspace/bionemo/data/cellxgene_2023-12-15/processed_data/test
dataset_path: /workspace/bionemo/data/singlecell_inf_tutorial/processed_data
dataset: /
data_prefix: ''
shuffle: true
medians_file: nemo:233c5b07146e47c78faf9883f28a99d0_medians.json
index_mapping_dir: null
skip_warmup: true
index_mapping_type: memmap
num_workers: 4
dataloader_type: single
seq_length: 2048
seed: 1234
dynamic_padding: true
micro_batch_size: 16
batch_size: 8
output_fname: /workspace/bionemo/data/singlecell_inf_tutorial/inference_output/human_covid19_bcells_pretrained_inference.pkl
data_fields_map:
sequence: sequence
id: id
data_impl_kwargs:
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
optim:
name: fused_adam
lr: 0.001
weight_decay: 0.1
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: 500
constant_steps: 2500
max_steps: 115430
min_lr: 2.0e-05
global_batch_size: 8
precision: bf16-mixed
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
nemo_version: 1.22.0
downstream_task:
restore_from_path: /workspace/bionemo/models/singlecell/geneformer/geneformer-106M-240530.nemo
outputs:
- embeddings
- hiddens
inference_output_everything: false
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
infer_target: bionemo.model.singlecell.geneformer.infer.GeneformerInference
formatters:
simple:
format: '[%(asctime)s][%(name)s][%(levelname)s] - %(message)s'
handlers:
console:
class: logging.StreamHandler
formatter: simple
stream: ext://sys.stdout
file:
class: logging.FileHandler
formatter: simple
filename: /logs/inference.log
root:
level: INFO
handlers:
- console
disable_existing_loggers: false
[NeMo W 2024-05-13 16:53:15 modelPT:251] You tried to register an artifact under config key=tokenizer.vocab_file but an artifact for it has already been registered.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: pipeline_model_parallel_split_rank in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo I 2024-05-13 16:53:15 megatron_init:234] Rank 0 has data parallel group: [0]
[NeMo I 2024-05-13 16:53:15 megatron_init:237] All data parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:15 megatron_init:238] Ranks 0 has data parallel rank: 0
[NeMo I 2024-05-13 16:53:15 megatron_init:246] Rank 0 has model parallel group: [0]
[NeMo I 2024-05-13 16:53:15 megatron_init:247] All model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:15 megatron_init:257] Rank 0 has tensor model parallel group: [0]
[NeMo I 2024-05-13 16:53:15 megatron_init:261] All tensor model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:15 megatron_init:262] Rank 0 has tensor model parallel rank: 0
[NeMo I 2024-05-13 16:53:15 megatron_init:276] Rank 0 has pipeline model parallel group: [0]
[NeMo I 2024-05-13 16:53:15 megatron_init:288] Rank 0 has embedding group: [0]
[NeMo I 2024-05-13 16:53:15 megatron_init:294] All pipeline model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:15 megatron_init:295] Rank 0 has pipeline model parallel rank 0
[NeMo I 2024-05-13 16:53:15 megatron_init:296] All embedding group ranks: [[0]]
[NeMo I 2024-05-13 16:53:15 megatron_init:297] Rank 0 has embedding rank: 0
24-05-13 16:53:15 - PID:289103 - rank:(0, 0, 0, 0) - microbatches.py:39 - INFO - setting number of micro-batches to constant 1
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: pipeline_model_parallel_split_rank in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 megatron_base_model:821] The model: GeneformerModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:15 modelPT:251] You tried to register an artifact under config key=tokenizer.vocab_file but an artifact for it has already been registered.
[NeMo I 2024-05-13 16:53:15 megatron_base_model:315] Padded vocab_size: 25472, original vocab_size: 25429, dummy tokens: 43.
[NeMo I 2024-05-13 16:53:16 nlp_overrides:752] Model GeneformerModel was successfully restored from /workspace/bionemo/models/singlecell/geneformer/geneformer-106M-240530.nemo.
[NeMo I 2024-05-13 16:53:16 utils:471] DDP is not initialized. Initializing...
Initializing distributed: GLOBAL_RANK: 0, MEMBER: 1/1
----------------------------------------------------------------------------------------------------
distributed_backend=nccl
All distributed processes registered. Starting with 1 processes
----------------------------------------------------------------------------------------------------
[NeMo W 2024-05-13 16:53:16 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/apex/transformer/pipeline_parallel/utils.py:81: UserWarning: This function is only for unittest
warnings.warn("This function is only for unittest")
[NeMo W 2024-05-13 16:53:16 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/modules/common/megatron/fused_bias_dropout_add.py:70: UserWarning: nvfuser integration in TorchScript is deprecated. (Triggered internally at /opt/pytorch/pytorch/torch/csrc/jit/codegen/cuda/interface.cpp:235.)
return bias_dropout_add_fused_inference_(*args)
[NeMo I 2024-05-13 16:53:17 loading:43]
************** Restored model configuration ***********
[NeMo I 2024-05-13 16:53:17 loading:44]
tokenizer:
vocab_file: /tmp/tmp_hpmn6eh/e0c840a303ab4ce7a8ad36400266741d_geneformer.vocab
micro_batch_size: 8
activation: relu
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
use_flash_attention: true
seq_length: 2048
encoder_seq_length: 2048
max_position_embeddings: 2048
num_layers: 12
hidden_size: 768
ffn_hidden_size: 3072
num_attention_heads: 12
init_method_std: 0.02
hidden_dropout: 0.02
attention_dropout: 0.02
kv_channels: null
apply_query_key_layer_scaling: true
layernorm_epsilon: 1.0e-12
make_vocab_size_divisible_by: 128
pre_process: true
post_process: false
bert_binary_head: false
resume_from_checkpoint: null
masked_softmax_fusion: true
native_amp_init_scale: 4294967296
native_amp_growth_interval: 1000
fp32_residual_connection: true
fp16_lm_cross_entropy: false
seed: 1234
use_cpu_initialization: false
onnx_safe: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
data:
data_impl: geneformer
probabilistic_dirichlet_sampling_train: false
train_dataset_path: /workspace/bionemo/data/cellxgene_2023-12-15/processed_data/train
val_dataset_path: /workspace/bionemo/data/cellxgene_2023-12-15/processed_data/val
test_dataset_path: /workspace/bionemo/data/cellxgene_2023-12-15/processed_data/test
dataset_path: /workspace/bionemo/data/singlecell_inf_tutorial/processed_data
dataset: /
data_prefix: ''
shuffle: true
medians_file: nemo:233c5b07146e47c78faf9883f28a99d0_medians.json
index_mapping_dir: null
skip_warmup: true
index_mapping_type: memmap
num_workers: 4
dataloader_type: single
seq_length: 2048
seed: 1234
dynamic_padding: true
micro_batch_size: 16
batch_size: 8
output_fname: /workspace/bionemo/data/singlecell_inf_tutorial/inference_output/human_covid19_bcells_pretrained_inference.pkl
data_fields_map:
sequence: sequence
id: id
data_impl_kwargs:
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
optim:
name: fused_adam
lr: 0.001
weight_decay: 0.1
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: 500
constant_steps: 2500
max_steps: 115430
min_lr: 2.0e-05
global_batch_size: 8
precision: bf16-mixed
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
nemo_version: 1.22.0
downstream_task:
restore_from_path: /workspace/bionemo/models/singlecell/geneformer/geneformer-106M-240530.nemo
outputs:
- embeddings
- hiddens
inference_output_everything: false
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0,1]
[NeMo W 2024-05-13 16:53:17 nemo_logging:349] /usr/lib/python3.10/multiprocessing/popen_fork.py:66: RuntimeWarning: os.fork() was called. os.fork() is incompatible with multithreaded code, and JAX is multithreaded, so this will likely lead to a deadlock.
self.pid = os.fork()
Predicting DataLoader 0: 100%|████████████████| 290/290 [01:23<00:00, 3.48it/s]
[NeMo I 2024-05-13 16:54:44 run_inference:50] Collecting results from all GPUs...
[NeMo I 2024-05-13 16:54:44 infer:73] Saving 2313 samples to /workspace/bionemo/data/singlecell_inf_tutorial/inference_output/human_covid19_bcells_pretrained_inference.pkl
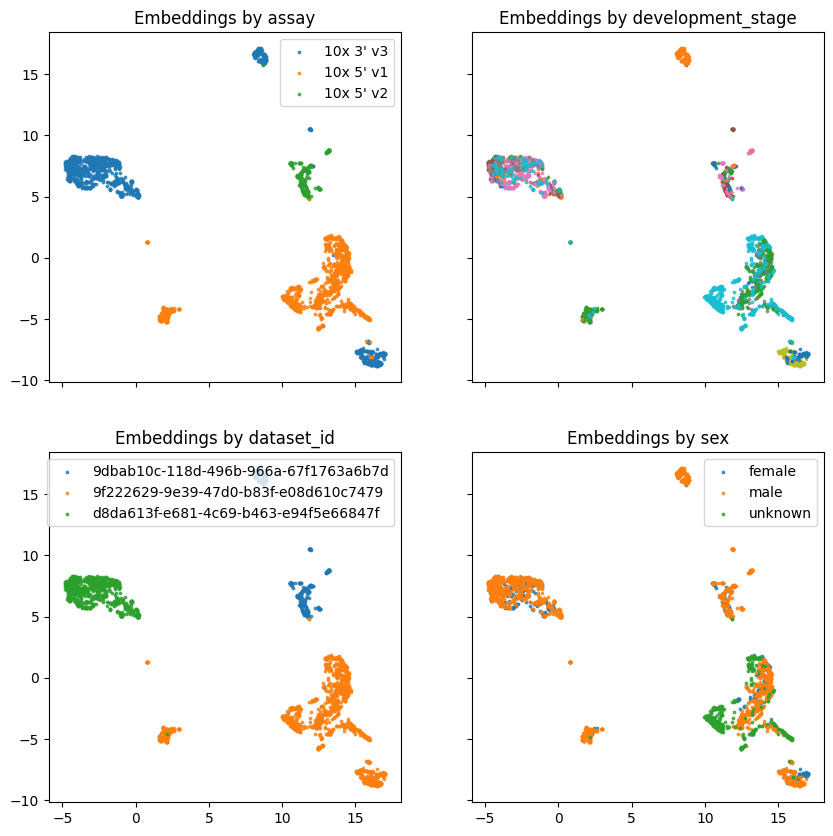
Load inference result and cluster with UMAP.#
Now we will inspect our result. First, we expect there to be one prediction for each cell, we can compare the shape of the anndata object to the predictions produced by our model. After this, we can simply pass our embeddings into umap, and view the result! In this case its a very poorly trained model with very few cells, so keep expectations low!
The inference_results pickle file contains one set of hiddens and embeddings for each cell. The hiddens contain the embedding per-token, whereas the embeddings contain the mean embedding for all gene tokens with special tokens (CLS, MASK, etc) removed.
import pickle
with open(tutorial_output_inference_pickle, 'rb') as inference_handle:
inference_results = pickle.load(inference_handle)
len(inference_results), inference_results[0].keys()
(2313, dict_keys(['embeddings']))
inference_results[0]['embeddings'].shape
(768,)
import umap
reducer = umap.UMAP()
embedding = reducer.fit_transform([x['embeddings'] for x in inference_results])
embedding.shape
(2313, 2)
from matplotlib import pyplot as plt
results = adata.obs.copy()
results['x'] = embedding[:, 0]
results['y'] = embedding[:, 1]
covariates = ["assay", "development_stage", "dataset_id", "sex"]
fig, axes = plt.subplots(nrows=2, ncols=2, sharex=True, sharey=True, figsize=(10,10))
for ax,covar in zip(axes.flat, covariates):
for cov, cov_df in results.groupby(covar):
ax.scatter(
cov_df.x,
cov_df.y,
s=3,
alpha=0.75,
label=cov,
)
if len(results[covar].unique()) < 8:
ax.legend()
ax.set_title(f"Embeddings by {covar}")