BioNeMo - Geneformer inferencing for single cell downstream tasks
Contents
BioNeMo - Geneformer inferencing for single cell downstream tasks#
This tutorial showcases how to run the BioNeMo container, pre-train a geneformer model, and use it for inferencing downstream single cell tasks. At the end of this tutorial, a user will learn:
launching the BioNeMo container
Download data from czi to use for pre-training and inference.
Convert AnnData files into the sparse CSR memmap format used by BioNeMo
Kick-off pretraining with a custom single cell dataset
Restore the pre-trained model and perform inference with the same czi dataset.
Prerequisites:#
BioNeMo Framework container is running (refer to the Quickstart Guide)
Familiarity with some components of the BioNeMo framework such as the Models and Inferencing
Running the BioNeMo container#
This example has been built by launching the container in a local machine with 2 x A6000 RTX GPUs. Refer to specific instructions for [remote and multi-node launch]
Once the container is launched, navigate to http://0.0.0.0:8888, http://localhost:8888, or the IP address of the workstation/node. A JupyterLab instance should show up.
Copy this code and input files into JupyterLab#
In the launched JupyterLab, run the codes in a Jupyter notebook as provided in the code cells below.
Getting example single cell data and setting it up for inference#
First, we must acquire single cell training data for inference. To do this we will install the cellxgene-census api and download a small dataset. We use the example provided by the czi api examples page to download a single h5ad file. Generally, our workflow expects a collection of h5ad files to be used for pre-training. In this case, we restrict to 100k cells from a single dataset to keep training time and downloading time small.
!pip install cellxgene-census
Defaulting to user installation because normal site-packages is not writeable
Looking in indexes: https://pypi.org/simple, https://pypi.ngc.nvidia.com
Requirement already satisfied: cellxgene-census in /workspace/bionemo/.local/lib/python3.10/site-packages (1.13.0)
Requirement already satisfied: tiledbsoma~=1.9.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from cellxgene-census) (1.9.5)
Requirement already satisfied: anndata in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (0.10.6)
Requirement already satisfied: numpy>=1.21 in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (1.24.4)
Requirement already satisfied: requests in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (2.31.0)
Requirement already satisfied: typing-extensions in /usr/local/lib/python3.10/dist-packages (from cellxgene-census) (4.7.1)
Requirement already satisfied: s3fs>=2021.06.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from cellxgene-census) (2024.3.1)
Requirement already satisfied: aiobotocore<3.0.0,>=2.5.4 in /workspace/bionemo/.local/lib/python3.10/site-packages (from s3fs>=2021.06.1->cellxgene-census) (2.12.3)
Requirement already satisfied: fsspec==2024.3.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from s3fs>=2021.06.1->cellxgene-census) (2024.3.1)
Requirement already satisfied: aiohttp!=4.0.0a0,!=4.0.0a1 in /usr/local/lib/python3.10/dist-packages (from s3fs>=2021.06.1->cellxgene-census) (3.9.0)
Requirement already satisfied: attrs>=22.2 in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (23.1.0)
Requirement already satisfied: numba>=0.58.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from tiledbsoma~=1.9.1->cellxgene-census) (0.59.1)
Requirement already satisfied: pandas in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.5.3)
Requirement already satisfied: pyarrow-hotfix in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (0.6)
Requirement already satisfied: scanpy>=1.9.2 in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.9.8)
Requirement already satisfied: scipy in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.11.1)
Requirement already satisfied: somacore==1.0.10 in /workspace/bionemo/.local/lib/python3.10/site-packages (from tiledbsoma~=1.9.1->cellxgene-census) (1.0.10)
Requirement already satisfied: tiledb~=0.27.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from tiledbsoma~=1.9.1->cellxgene-census) (0.27.1)
Requirement already satisfied: pyarrow>=9.0.0 in /usr/local/lib/python3.10/dist-packages (from tiledbsoma~=1.9.1->cellxgene-census) (14.0.1)
Requirement already satisfied: array-api-compat!=1.5,>1.4 in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (1.5.1)
Requirement already satisfied: exceptiongroup in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (1.1.3)
Requirement already satisfied: h5py>=3.1 in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (3.10.0)
Requirement already satisfied: natsort in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (8.4.0)
Requirement already satisfied: packaging>=20.0 in /usr/local/lib/python3.10/dist-packages (from anndata->cellxgene-census) (23.1)
Requirement already satisfied: charset-normalizer<4,>=2 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (3.2.0)
Requirement already satisfied: idna<4,>=2.5 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (3.4)
Requirement already satisfied: urllib3<3,>=1.21.1 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (1.26.16)
Requirement already satisfied: certifi>=2017.4.17 in /usr/local/lib/python3.10/dist-packages (from requests->cellxgene-census) (2023.7.22)
Requirement already satisfied: botocore<1.34.70,>=1.34.41 in /workspace/bionemo/.local/lib/python3.10/site-packages (from aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (1.34.69)
Requirement already satisfied: wrapt<2.0.0,>=1.10.10 in /usr/local/lib/python3.10/dist-packages (from aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (1.14.1)
Requirement already satisfied: aioitertools<1.0.0,>=0.5.1 in /workspace/bionemo/.local/lib/python3.10/site-packages (from aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (0.11.0)
Requirement already satisfied: multidict<7.0,>=4.5 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (6.0.4)
Requirement already satisfied: yarl<2.0,>=1.0 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (1.9.2)
Requirement already satisfied: frozenlist>=1.1.1 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (1.4.0)
Requirement already satisfied: aiosignal>=1.1.2 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (1.3.1)
Requirement already satisfied: async-timeout<5.0,>=4.0 in /usr/local/lib/python3.10/dist-packages (from aiohttp!=4.0.0a0,!=4.0.0a1->s3fs>=2021.06.1->cellxgene-census) (4.0.3)
Requirement already satisfied: llvmlite<0.43,>=0.42.0dev0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from numba>=0.58.0->tiledbsoma~=1.9.1->cellxgene-census) (0.42.0)
Requirement already satisfied: python-dateutil>=2.8.1 in /usr/local/lib/python3.10/dist-packages (from pandas->tiledbsoma~=1.9.1->cellxgene-census) (2.8.2)
Requirement already satisfied: pytz>=2020.1 in /usr/local/lib/python3.10/dist-packages (from pandas->tiledbsoma~=1.9.1->cellxgene-census) (2023.3)
Requirement already satisfied: joblib in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.2.0)
Requirement already satisfied: matplotlib>=3.6 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (3.8.0)
Requirement already satisfied: networkx>=2.3 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (2.6.3)
Requirement already satisfied: patsy in /workspace/bionemo/.local/lib/python3.10/site-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.5.6)
Requirement already satisfied: scikit-learn>=0.24 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.2.0)
Requirement already satisfied: seaborn>=0.13.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.13.2)
Requirement already satisfied: session-info in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.0.0)
Requirement already satisfied: statsmodels>=0.10.0rc2 in /workspace/bionemo/.local/lib/python3.10/site-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.14.1)
Requirement already satisfied: tqdm in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (4.66.1)
Requirement already satisfied: umap-learn>=0.3.10 in /usr/local/lib/python3.10/dist-packages (from scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.5.5)
Requirement already satisfied: jmespath<2.0.0,>=0.7.1 in /usr/local/lib/python3.10/dist-packages (from botocore<1.34.70,>=1.34.41->aiobotocore<3.0.0,>=2.5.4->s3fs>=2021.06.1->cellxgene-census) (1.0.1)
Requirement already satisfied: contourpy>=1.0.1 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.1.1)
Requirement already satisfied: cycler>=0.10 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.12.1)
Requirement already satisfied: fonttools>=4.22.0 in /workspace/bionemo/.local/lib/python3.10/site-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (4.51.0)
Requirement already satisfied: kiwisolver>=1.0.1 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (1.4.5)
Requirement already satisfied: pillow>=6.2.0 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (10.2.0)
Requirement already satisfied: pyparsing>=2.3.1 in /usr/local/lib/python3.10/dist-packages (from matplotlib>=3.6->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (3.1.1)
Requirement already satisfied: six>=1.5 in /usr/local/lib/python3.10/dist-packages (from python-dateutil>=2.8.1->pandas->tiledbsoma~=1.9.1->cellxgene-census) (1.16.0)
Requirement already satisfied: threadpoolctl>=2.0.0 in /usr/local/lib/python3.10/dist-packages (from scikit-learn>=0.24->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (3.2.0)
Requirement already satisfied: pynndescent>=0.5 in /usr/local/lib/python3.10/dist-packages (from umap-learn>=0.3.10->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.5.11)
Requirement already satisfied: stdlib-list in /usr/local/lib/python3.10/dist-packages (from session-info->scanpy>=1.9.2->tiledbsoma~=1.9.1->cellxgene-census) (0.10.0)
[notice] A new release of pip is available: 23.2.1 -> 24.0
[notice] To update, run: python -m pip install --upgrade pip
# Below are paths required for setting up pre-training and inference.
tutorial_data_dir = "/workspace/bionemo/data/singlecell_tutorial/download_anndata"
train_tutorial_data_dir = "/workspace/bionemo/data/singlecell_tutorial/download_anndata/train"
val_tutorial_data_dir = "/workspace/bionemo/data/singlecell_tutorial/download_anndata/val"
test_tutorial_data_dir = "/workspace/bionemo/data/singlecell_tutorial/download_anndata/test"
train_tutorial_processed_dir = "/workspace/bionemo/data/singlecell_tutorial/processed_data/train"
val_tutorial_processed_dir = "/workspace/bionemo/data/singlecell_tutorial/processed_data/val"
test_tutorial_processed_dir = "/workspace/bionemo/data/singlecell_tutorial/processed_data/test"
tutorial_output_dir = "/workspace/bionemo/data/singlecell_tutorial/inference_output"
tutorial_output_inference_pickle = f"{tutorial_output_dir}/human_covid19_bcells_from_scratch.pkl"
demo_data_train_download_path = f"{train_tutorial_data_dir}/human_covid19_bcells.h5ad"
demo_data_val_download_path = f"{val_tutorial_data_dir}/human_covid19_bcells.h5ad"
demo_data_test_download_path = f"{test_tutorial_data_dir}/human_covid19_bcells.h5ad"
!mkdir -p {train_tutorial_data_dir}
!mkdir -p {val_tutorial_data_dir}
!mkdir -p {test_tutorial_data_dir}
!mkdir -p {train_tutorial_processed_dir}
!mkdir -p {val_tutorial_processed_dir}
!mkdir -p {test_tutorial_processed_dir}
!mkdir -p {tutorial_output_dir}
import cellxgene_census
frac_train = 0.8
frac_val = 0.1
frac_test = 0.1
with cellxgene_census.open_soma(census_version="2023-12-15") as census:
filter1 = "cell_type == 'B cell' and tissue_general == 'lung' and disease == 'COVID-19' and is_primary_data == True"
adata = cellxgene_census.get_anndata(
census = census,
organism = "Homo sapiens",
obs_value_filter = filter1,
)
n_train = int(adata.shape[0] * frac_train)
n_val = int(adata.shape[0] * frac_val)
n_test = adata.shape[0] - n_train - n_val
# Create some splits, bad practice since ordering may be a thing but let's just take ranges for this demo.
adata_train = adata[0:n_train].copy()
adata_val = adata[n_train:(n_train+n_val)].copy()
adata_test = adata[(n_train+n_val):].copy()
adata_train.write(demo_data_train_download_path)
adata_val.write(demo_data_val_download_path)
adata_test.write(demo_data_test_download_path)
!ls -laht {demo_data_download_path}
ls: cannot access '{demo_data_download_path}': No such file or directory
# Create training data processed directory
!python /workspace/bionemo/bionemo/data/singlecell/sc_memmap.py \
--data-path {train_tutorial_data_dir} \
--save-path {train_tutorial_processed_dir}
# Create validation data processed directory
!python /workspace/bionemo/bionemo/data/singlecell/sc_memmap.py \
--data-path {val_tutorial_data_dir} \
--save-path {val_tutorial_processed_dir}
# Create test data processed directory
!python /workspace/bionemo/bionemo/data/singlecell/sc_memmap.py \
--data-path {test_tutorial_data_dir} \
--save-path {test_tutorial_processed_dir}
Found 1 files
Starting to create memmap files...
Creating metadata...: 100%|███████████████████████| 1/1 [00:00<00:00, 9.13it/s]
Done creating `metadata.json`
Writing data into memmaps to /workspace/bionemo/data/singlecell_tutorial/processed_data/train...
Merging AnnData into numpy memaps...: 100%|███████| 1/1 [00:00<00:00, 6.56it/s]
Saving dataframe ...
Done creating dataset ...
Found 1 files
Starting to create memmap files...
Creating metadata...: 100%|███████████████████████| 1/1 [00:00<00:00, 9.86it/s]
Done creating `metadata.json`
Writing data into memmaps to /workspace/bionemo/data/singlecell_tutorial/processed_data/val...
Merging AnnData into numpy memaps...: 100%|███████| 1/1 [00:00<00:00, 8.41it/s]
Saving dataframe ...
Done creating dataset ...
Found 1 files
Starting to create memmap files...
Creating metadata...: 100%|███████████████████████| 1/1 [00:00<00:00, 9.72it/s]
Done creating `metadata.json`
Writing data into memmaps to /workspace/bionemo/data/singlecell_tutorial/processed_data/test...
Merging AnnData into numpy memaps...: 100%|███████| 1/1 [00:00<00:00, 8.02it/s]
Saving dataframe ...
Done creating dataset ...
!ls -laht {train_tutorial_processed_dir}
total 13M
-rw-r--r-- 1 jstjohn domain-users 157K May 13 16:52 features.csv
drwxr-xr-x 2 jstjohn domain-users 4.0K May 13 16:52 .
-rw-r--r-- 1 jstjohn domain-users 5.9M May 13 16:52 gene_expression_ind.npy
-rw-r--r-- 1 jstjohn domain-users 15K May 13 16:52 gene_expression_ptr.npy
-rw-r--r-- 1 jstjohn domain-users 5.9M May 13 16:52 gene_expression_data.npy
-rw-r--r-- 1 jstjohn domain-users 1.1M May 13 16:52 metadata.json
drwxr-xr-x 5 jstjohn domain-users 4.0K May 13 16:49 ..
Pretraining#
Now that we have aquired the h5ad files we would like to use for training and converted them to a sparse memmap. We will kickoff training. This involves two distinct steps
preprocessing (indicated with do_training=False), where artifacts are downloaded from huggingface to be used by the model. Importantly, we set the
dataset_pathto be the same place we created the sparse memmap files. This is how BioNeMo knows where to find files for training, including both training data and additional artifacts (such as tokenizers).pretraining, where the model is actually trained.
We set the flag max_steps to limit the runtime. Check the full config file in examples/singlecell/geneformer/conf for a complete list of arguments.
# Run preprocessing to acquire the requisite files for pre-training.
!python /workspace/bionemo/examples/singlecell/geneformer/pretrain.py \
++model.data.train_dataset_path={train_tutorial_processed_dir} \
++model.data.val_dataset_path={val_tutorial_processed_dir} \
++model.data.test_dataset_path={test_tutorial_processed_dir} \
++do_training=False
[NeMo W 2024-05-13 16:53:05 nemo_logging:349] /usr/lib/python3.10/multiprocessing/popen_fork.py:66: RuntimeWarning: os.fork() was called. os.fork() is incompatible with multithreaded code, and JAX is multithreaded, so this will likely lead to a deadlock.
self.pid = os.fork()
[NeMo I 2024-05-13 16:53:07 megatron_hiddens:110] Registered hidden transform sampled_var_cond_gaussian at bionemo.model.core.hiddens_support.SampledVarGaussianHiddenTransform
[NeMo I 2024-05-13 16:53:07 megatron_hiddens:110] Registered hidden transform interp_var_cond_gaussian at bionemo.model.core.hiddens_support.InterpVarGaussianHiddenTransform
[NeMo W 2024-05-13 16:53:07 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/hydra/_internal/defaults_list.py:251: UserWarning: In 'geneformer_config': Defaults list is missing `_self_`. See https://hydra.cc/docs/1.2/upgrades/1.0_to_1.1/default_composition_order for more information
warnings.warn(msg, UserWarning)
[NeMo W 2024-05-13 16:53:07 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/hydra/_internal/hydra.py:119: UserWarning: Future Hydra versions will no longer change working directory at job runtime by default.
See https://hydra.cc/docs/1.2/upgrades/1.1_to_1.2/changes_to_job_working_dir/ for more information.
ret = run_job(
[NeMo I 2024-05-13 16:53:07 pretrain:33]
************** Experiment configuration ***********
[NeMo I 2024-05-13 16:53:07 pretrain:34]
name: geneformer_base_config
restore_from_path: null
seed_everything: false
do_training: false
trainer:
devices: 1
num_nodes: 1
accelerator: gpu
precision: bf16-mixed
logger: false
enable_checkpointing: false
use_distributed_sampler: false
max_epochs: 1
max_steps: 400000
log_every_n_steps: 100
val_check_interval: 100
limit_val_batches: 8
limit_test_batches: 500
accumulate_grad_batches: 1
gradient_clip_val: 1.0
benchmark: false
exp_manager:
explicit_log_dir: null
exp_dir: ${oc.env:BIONEMO_HOME}/results/nemo_experiments/${.name}/${.wandb_logger_kwargs.name}
name: geneformer
create_wandb_logger: true
wandb_logger_kwargs:
project: null
name: geneformer-pretraining
offline: false
resume_if_exists: true
resume_ignore_no_checkpoint: true
create_checkpoint_callback: true
checkpoint_callback_params:
monitor: val_loss
save_top_k: 1
mode: min
always_save_nemo: true
filename: geneformer--{val_loss:.2f}-{step}-{consumed_samples}
model_parallel_size: ${multiply:${model.tensor_model_parallel_size}, ${model.pipeline_model_parallel_size}}
model:
tokenizer:
vocab_file: ${..data.train_dataset_path}/geneformer.vocab
micro_batch_size: 8
activation: relu
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
use_flash_attention: true
seq_length: 2048
encoder_seq_length: ${.seq_length}
max_position_embeddings: ${.seq_length}
num_layers: 6
hidden_size: 256
ffn_hidden_size: 512
num_attention_heads: 4
init_method_std: 0.02
hidden_dropout: 0.02
attention_dropout: 0.02
kv_channels: null
apply_query_key_layer_scaling: true
layernorm_epsilon: 1.0e-12
make_vocab_size_divisible_by: 128
pre_process: true
post_process: true
bert_binary_head: false
resume_from_checkpoint: null
masked_softmax_fusion: true
native_amp_init_scale: 4294967296
native_amp_growth_interval: 1000
fp32_residual_connection: true
fp16_lm_cross_entropy: false
seed: 1234
use_cpu_initialization: false
onnx_safe: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
data:
data_impl: geneformer
probabilistic_dirichlet_sampling_train: false
train_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/train
val_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/val
test_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
dataset_path: null
dataset: /
data_prefix: ''
shuffle: true
medians_file: ${.train_dataset_path}/medians.json
index_mapping_dir: ${exp_manager.exp_dir}/index_mapping
skip_warmup: true
index_mapping_type: memmap
num_workers: 12
dataloader_type: single
seq_length: ${model.seq_length}
seed: ${model.seed}
dynamic_padding: true
micro_batch_size: ${model.micro_batch_size}
optim:
name: fused_adam
lr: 0.001
weight_decay: 0.1
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: ${multiply:${trainer.max_steps}, 0.01}
constant_steps: ${multiply:${trainer.max_steps}, 0.05}
max_steps: ${trainer.max_steps}
min_lr: 2.0e-05
[NeMo I 2024-05-13 16:53:07 pretrain:51] ************** Starting Preprocessing ***********
[NeMo I 2024-05-13 16:53:07 remote:103] Downloading resource: https://huggingface.co/ctheodoris/Geneformer/resolve/main/geneformer/gene_name_id_dict.pkl?download=true
[NeMo I 2024-05-13 16:53:07 remote:121] No checksum provided, filename exists. Assuming it is complete.
[NeMo I 2024-05-13 16:53:07 remote:103] Downloading resource: https://huggingface.co/ctheodoris/Geneformer/resolve/main/geneformer/gene_median_dictionary.pkl?download=true
[NeMo I 2024-05-13 16:53:08 remote:121] No checksum provided, filename exists. Assuming it is complete.
[NeMo I 2024-05-13 16:53:08 pretrain:60] *************** Preprocessing Finished ************
# Pretrain the model using
!python /workspace/bionemo/examples/singlecell/geneformer/pretrain.py \
--config-dir /workspace/bionemo/examples/singlecell/geneformer/conf \
--config-name geneformer_config \
++model.data.train_dataset_path={train_tutorial_processed_dir} \
++model.data.val_dataset_path={val_tutorial_processed_dir} \
++model.data.test_dataset_path={test_tutorial_processed_dir} \
++trainer.devices=1 \
++trainer.max_steps=200 \
++exp_manager.exp_dir={tutorial_output_dir} \
++exp_manager.wandb_logger_kwargs.project="geneformer_pretrain_test" \
++model.data.output_fname={tutorial_output_inference_pickle} \
++exp_manager.wandb_logger_kwargs.offline=True \
++exp_manager.resume_if_exists=False \
++do_training=True
[NeMo W 2024-05-13 16:53:22 nemo_logging:349] /usr/lib/python3.10/multiprocessing/popen_fork.py:66: RuntimeWarning: os.fork() was called. os.fork() is incompatible with multithreaded code, and JAX is multithreaded, so this will likely lead to a deadlock.
self.pid = os.fork()
[NeMo I 2024-05-13 16:53:24 megatron_hiddens:110] Registered hidden transform sampled_var_cond_gaussian at bionemo.model.core.hiddens_support.SampledVarGaussianHiddenTransform
[NeMo I 2024-05-13 16:53:24 megatron_hiddens:110] Registered hidden transform interp_var_cond_gaussian at bionemo.model.core.hiddens_support.InterpVarGaussianHiddenTransform
[NeMo W 2024-05-13 16:53:24 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/hydra/_internal/defaults_list.py:251: UserWarning: In 'geneformer_config': Defaults list is missing `_self_`. See https://hydra.cc/docs/1.2/upgrades/1.0_to_1.1/default_composition_order for more information
warnings.warn(msg, UserWarning)
[NeMo W 2024-05-13 16:53:24 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/hydra/_internal/hydra.py:119: UserWarning: Future Hydra versions will no longer change working directory at job runtime by default.
See https://hydra.cc/docs/1.2/upgrades/1.1_to_1.2/changes_to_job_working_dir/ for more information.
ret = run_job(
[NeMo I 2024-05-13 16:53:24 pretrain:33]
************** Experiment configuration ***********
[NeMo I 2024-05-13 16:53:24 pretrain:34]
name: geneformer_base_config
restore_from_path: null
seed_everything: false
do_training: true
trainer:
devices: 1
num_nodes: 1
accelerator: gpu
precision: bf16-mixed
logger: false
enable_checkpointing: false
use_distributed_sampler: false
max_epochs: 1
max_steps: 200
log_every_n_steps: 100
val_check_interval: 100
limit_val_batches: 8
limit_test_batches: 500
accumulate_grad_batches: 1
gradient_clip_val: 1.0
benchmark: false
exp_manager:
explicit_log_dir: null
exp_dir: /workspace/bionemo/data/singlecell_tutorial/inference_output
name: geneformer
create_wandb_logger: true
wandb_logger_kwargs:
project: geneformer_pretrain_test
name: geneformer-pretraining
offline: true
resume_if_exists: false
resume_ignore_no_checkpoint: true
create_checkpoint_callback: true
checkpoint_callback_params:
monitor: val_loss
save_top_k: 1
mode: min
always_save_nemo: true
filename: geneformer--{val_loss:.2f}-{step}-{consumed_samples}
model_parallel_size: ${multiply:${model.tensor_model_parallel_size}, ${model.pipeline_model_parallel_size}}
model:
tokenizer:
vocab_file: ${..data.train_dataset_path}/geneformer.vocab
micro_batch_size: 8
activation: relu
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
use_flash_attention: true
seq_length: 2048
encoder_seq_length: ${.seq_length}
max_position_embeddings: ${.seq_length}
num_layers: 6
hidden_size: 256
ffn_hidden_size: 512
num_attention_heads: 4
init_method_std: 0.02
hidden_dropout: 0.02
attention_dropout: 0.02
kv_channels: null
apply_query_key_layer_scaling: true
layernorm_epsilon: 1.0e-12
make_vocab_size_divisible_by: 128
pre_process: true
post_process: true
bert_binary_head: false
resume_from_checkpoint: null
masked_softmax_fusion: true
native_amp_init_scale: 4294967296
native_amp_growth_interval: 1000
fp32_residual_connection: true
fp16_lm_cross_entropy: false
seed: 1234
use_cpu_initialization: false
onnx_safe: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
data:
data_impl: geneformer
probabilistic_dirichlet_sampling_train: false
train_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/train
val_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/val
test_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
dataset_path: null
dataset: /
data_prefix: ''
shuffle: true
medians_file: ${.train_dataset_path}/medians.json
index_mapping_dir: ${exp_manager.exp_dir}/index_mapping
skip_warmup: true
index_mapping_type: memmap
num_workers: 12
dataloader_type: single
seq_length: ${model.seq_length}
seed: ${model.seed}
dynamic_padding: true
micro_batch_size: ${model.micro_batch_size}
output_fname: /workspace/bionemo/data/singlecell_tutorial/inference_output/human_covid19_bcells_from_scratch.pkl
optim:
name: fused_adam
lr: 0.001
weight_decay: 0.1
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: ${multiply:${trainer.max_steps}, 0.01}
constant_steps: ${multiply:${trainer.max_steps}, 0.05}
max_steps: ${trainer.max_steps}
min_lr: 2.0e-05
[NeMo I 2024-05-13 16:53:24 utils:230] Selected Callbacks: [<class 'pytorch_lightning.callbacks.model_summary.ModelSummary'>]
Trainer already configured with model summary callbacks: [<class 'pytorch_lightning.callbacks.model_summary.ModelSummary'>]. Skipping setting a default `ModelSummary` callback.
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
HPU available: False, using: 0 HPUs
[NeMo I 2024-05-13 16:53:24 exp_manager:394] Experiments will be logged at /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24
[NeMo I 2024-05-13 16:53:24 exp_manager:835] TensorboardLogger has been set up
wandb: WARNING `resume` will be ignored since W&B syncing is set to `offline`. Starting a new run with run id 2024-05-13_16-53-24.
wandb: Tracking run with wandb version 0.15.6
wandb: W&B syncing is set to `offline` in this directory.
wandb: Run `wandb online` or set WANDB_MODE=online to enable cloud syncing.
[NeMo I 2024-05-13 16:53:25 exp_manager:850] WandBLogger has been set up
[NeMo W 2024-05-13 16:53:25 exp_manager:931] The checkpoint callback was told to monitor a validation value and trainer's max_steps was set to 200. Please ensure that max_steps will run for at least 1 epochs to ensure that checkpointing will not error out.
[NeMo I 2024-05-13 16:53:25 utils:306]
************** Trainer configuration ***********
[NeMo I 2024-05-13 16:53:25 utils:307]
name: geneformer_base_config
restore_from_path: null
seed_everything: false
do_training: true
trainer:
devices: 1
num_nodes: 1
accelerator: gpu
precision: bf16-mixed
logger: false
enable_checkpointing: false
use_distributed_sampler: false
max_epochs: 1
max_steps: 200
log_every_n_steps: 100
val_check_interval: 100
limit_val_batches: 8
limit_test_batches: 500
accumulate_grad_batches: 1
gradient_clip_val: 1.0
benchmark: false
exp_manager:
explicit_log_dir: null
exp_dir: /workspace/bionemo/data/singlecell_tutorial/inference_output
name: geneformer
create_wandb_logger: true
wandb_logger_kwargs:
project: geneformer_pretrain_test
name: geneformer-pretraining
offline: true
resume_if_exists: false
resume_ignore_no_checkpoint: true
create_checkpoint_callback: true
checkpoint_callback_params:
monitor: val_loss
save_top_k: 1
mode: min
always_save_nemo: true
filename: geneformer--{val_loss:.2f}-{step}-{consumed_samples}
model_parallel_size: ${multiply:${model.tensor_model_parallel_size}, ${model.pipeline_model_parallel_size}}
model:
tokenizer:
vocab_file: ${..data.train_dataset_path}/geneformer.vocab
micro_batch_size: 8
activation: relu
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
use_flash_attention: true
seq_length: 2048
encoder_seq_length: ${.seq_length}
max_position_embeddings: ${.seq_length}
num_layers: 6
hidden_size: 256
ffn_hidden_size: 512
num_attention_heads: 4
init_method_std: 0.02
hidden_dropout: 0.02
attention_dropout: 0.02
kv_channels: null
apply_query_key_layer_scaling: true
layernorm_epsilon: 1.0e-12
make_vocab_size_divisible_by: 128
pre_process: true
post_process: true
bert_binary_head: false
resume_from_checkpoint: null
masked_softmax_fusion: true
native_amp_init_scale: 4294967296
native_amp_growth_interval: 1000
fp32_residual_connection: true
fp16_lm_cross_entropy: false
seed: 1234
use_cpu_initialization: false
onnx_safe: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
data:
data_impl: geneformer
probabilistic_dirichlet_sampling_train: false
train_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/train
val_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/val
test_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
dataset_path: null
dataset: /
data_prefix: ''
shuffle: true
medians_file: ${.train_dataset_path}/medians.json
index_mapping_dir: ${exp_manager.exp_dir}/index_mapping
skip_warmup: true
index_mapping_type: memmap
num_workers: 12
dataloader_type: single
seq_length: ${model.seq_length}
seed: ${model.seed}
dynamic_padding: true
micro_batch_size: ${model.micro_batch_size}
output_fname: /workspace/bionemo/data/singlecell_tutorial/inference_output/human_covid19_bcells_from_scratch.pkl
optim:
name: fused_adam
lr: 0.001
weight_decay: 0.1
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: ${multiply:${trainer.max_steps}, 0.01}
constant_steps: ${multiply:${trainer.max_steps}, 0.05}
max_steps: ${trainer.max_steps}
min_lr: 2.0e-05
global_batch_size: 8
precision: bf16-mixed
[NeMo W 2024-05-13 16:53:25 modelPT:251] You tried to register an artifact under config key=tokenizer.vocab_file but an artifact for it has already been registered.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: pipeline_model_parallel_split_rank in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo I 2024-05-13 16:53:25 megatron_init:234] Rank 0 has data parallel group: [0]
[NeMo I 2024-05-13 16:53:25 megatron_init:237] All data parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:25 megatron_init:238] Ranks 0 has data parallel rank: 0
[NeMo I 2024-05-13 16:53:25 megatron_init:246] Rank 0 has model parallel group: [0]
[NeMo I 2024-05-13 16:53:25 megatron_init:247] All model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:25 megatron_init:257] Rank 0 has tensor model parallel group: [0]
[NeMo I 2024-05-13 16:53:25 megatron_init:261] All tensor model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:25 megatron_init:262] Rank 0 has tensor model parallel rank: 0
[NeMo I 2024-05-13 16:53:25 megatron_init:276] Rank 0 has pipeline model parallel group: [0]
[NeMo I 2024-05-13 16:53:25 megatron_init:288] Rank 0 has embedding group: [0]
[NeMo I 2024-05-13 16:53:25 megatron_init:294] All pipeline model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:53:25 megatron_init:295] Rank 0 has pipeline model parallel rank 0
[NeMo I 2024-05-13 16:53:25 megatron_init:296] All embedding group ranks: [[0]]
[NeMo I 2024-05-13 16:53:25 megatron_init:297] Rank 0 has embedding rank: 0
24-05-13 16:53:25 - PID:290675 - rank:(0, 0, 0, 0) - microbatches.py:39 - INFO - setting number of micro-batches to constant 1
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: pipeline_model_parallel_split_rank in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 megatron_base_model:821] The model: GeneformerModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:53:25 modelPT:251] You tried to register an artifact under config key=tokenizer.vocab_file but an artifact for it has already been registered.
[NeMo I 2024-05-13 16:53:25 megatron_base_model:315] Padded vocab_size: 25472, original vocab_size: 25429, dummy tokens: 43.
[NeMo I 2024-05-13 16:53:26 pretrain:47] ************** Starting Training ***********
[NeMo W 2024-05-13 16:53:26 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/trainer/configuration_validator.py:153: UserWarning: The `batch_idx` argument in `GeneformerModel.on_train_batch_start` hook may not match with the actual batch index when using a `dataloader_iter` argument in your `training_step`.
rank_zero_warn(
[NeMo W 2024-05-13 16:53:26 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/trainer/configuration_validator.py:153: UserWarning: The `batch_idx` argument in `GeneformerModel.on_train_batch_end` hook may not match with the actual batch index when using a `dataloader_iter` argument in your `training_step`.
rank_zero_warn(
Initializing distributed: GLOBAL_RANK: 0, MEMBER: 1/1
----------------------------------------------------------------------------------------------------
distributed_backend=nccl
All distributed processes registered. Starting with 1 processes
----------------------------------------------------------------------------------------------------
Pipeline model parallel rank: 0, Tensor model parallel rank: 0, Number of model parameters on device: 1.03e+07. Total number of model parameters: 1.03e+07.
[NeMo I 2024-05-13 16:53:26 core:263] Building Bert datasets.
> WARNING: could not find index map file /workspace/bionemo/data/singlecell_tutorial/inference_output/index_mapping/_train_1600_indexmap_1600mns_2046msl_0.00ssp_1234s.npy, building the indices on rank 0 ...
[NeMo I 2024-05-13 16:53:26 dataset_utils:1303] > building samples index mapping for train_1600 ...
make: Entering directory '/usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/data/language_modeling/megatron'
make: Nothing to be done for 'default'.
make: Leaving directory '/usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/data/language_modeling/megatron'
using uint32 for data mapping...
using:
number of documents: 1850
sentences range: [0, 1850)
total number of sentences: 1850
number of epochs: 2147483646
maximum number of samples: 1600
maximum sequence length: 2046
short sequence probability: 0
short sequence ration (1/prob): 0
seed: 1234
reached 1600 samples after 1 epochs ...
number of empty documents: 0
number of documents with one sentence: 1850
number of documents with long sentences: 0
will create mapping for 1850 samples
[NeMo I 2024-05-13 16:53:26 dataset_utils:1324] > done building samples index maping
[NeMo I 2024-05-13 16:53:26 dataset_utils:1326] > saved the index mapping in /workspace/bionemo/data/singlecell_tutorial/inference_output/index_mapping/_train_1600_indexmap_1600mns_2046msl_0.00ssp_1234s.npy
[NeMo I 2024-05-13 16:53:26 dataset_utils:1328] > elasped time to build and save samples mapping (seconds): 0.050445
[NeMo W 2024-05-13 16:53:26 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/data/language_modeling/megatron/dataset_utils.py:1332: UserWarning: The torch.cuda.*DtypeTensor constructors are no longer recommended. It's best to use methods such as torch.tensor(data, dtype=*, device='cuda') to create tensors. (Triggered internally at /opt/pytorch/pytorch/torch/csrc/tensor/python_tensor.cpp:83.)
counts = torch.cuda.LongTensor([1])
> WARNING: could not find index map file /workspace/bionemo/data/singlecell_tutorial/inference_output/index_mapping/_val_64_indexmap_64mns_2046msl_0.00ssp_1234s.npy, building the indices on rank 0 ...
[NeMo I 2024-05-13 16:53:27 dataset_utils:1303] > building samples index mapping for val_64 ...
make: Entering directory '/usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/data/language_modeling/megatron'
make: Nothing to be done for 'default'.
make: Leaving directory '/usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/data/language_modeling/megatron'
using uint32 for data mapping...
using:
number of documents: 231
sentences range: [0, 231)
total number of sentences: 231
number of epochs: 2147483646
maximum number of samples: 64
maximum sequence length: 2046
short sequence probability: 0
short sequence ration (1/prob): 0
seed: 1234
reached 64 samples after 1 epochs ...
number of empty documents: 0
number of documents with one sentence: 231
number of documents with long sentences: 0
will create mapping for 231 samples
[NeMo I 2024-05-13 16:53:27 dataset_utils:1324] > done building samples index maping
[NeMo I 2024-05-13 16:53:27 dataset_utils:1326] > saved the index mapping in /workspace/bionemo/data/singlecell_tutorial/inference_output/index_mapping/_val_64_indexmap_64mns_2046msl_0.00ssp_1234s.npy
[NeMo I 2024-05-13 16:53:27 dataset_utils:1328] > elasped time to build and save samples mapping (seconds): 0.046324
> WARNING: could not find index map file /workspace/bionemo/data/singlecell_tutorial/inference_output/index_mapping/_test_232_indexmap_232mns_2046msl_0.00ssp_1234s.npy, building the indices on rank 0 ...
[NeMo I 2024-05-13 16:53:27 dataset_utils:1303] > building samples index mapping for test_232 ...
make: Entering directory '/usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/data/language_modeling/megatron'
make: Nothing to be done for 'default'.
make: Leaving directory '/usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/data/language_modeling/megatron'
using uint32 for data mapping...
using:
number of documents: 232
sentences range: [0, 232)
total number of sentences: 232
number of epochs: 2147483646
maximum number of samples: 232
maximum sequence length: 2046
short sequence probability: 0
short sequence ration (1/prob): 0
seed: 1234
reached 232 samples after 1 epochs ...
number of empty documents: 0
number of documents with one sentence: 232
number of documents with long sentences: 0
will create mapping for 232 samples
[NeMo I 2024-05-13 16:53:27 dataset_utils:1324] > done building samples index maping
[NeMo I 2024-05-13 16:53:27 dataset_utils:1326] > saved the index mapping in /workspace/bionemo/data/singlecell_tutorial/inference_output/index_mapping/_test_232_indexmap_232mns_2046msl_0.00ssp_1234s.npy
[NeMo I 2024-05-13 16:53:27 dataset_utils:1328] > elasped time to build and save samples mapping (seconds): 0.042735
[NeMo I 2024-05-13 16:53:27 core:269] Length of train dataset: 1600
[NeMo I 2024-05-13 16:53:27 core:270] Length of val dataset: 64
[NeMo I 2024-05-13 16:53:27 core:271] Length of test dataset: 232
[NeMo I 2024-05-13 16:53:27 core:272] Finished building Bert datasets.
Setting up train dataloader with len(len(self._train_ds)): 1600 and consumed samples: 0
[NeMo I 2024-05-13 16:53:27 data_samplers:76] Instantiating MegatronPretrainingSampler with total_samples: 1600 and consumed_samples: 0
[NeMo I 2024-05-13 16:53:27 data_samplers:76] Instantiating MegatronPretrainingSampler with total_samples: 64 and consumed_samples: 0
[NeMo I 2024-05-13 16:53:27 data_samplers:76] Instantiating MegatronPretrainingSampler with total_samples: 232 and consumed_samples: 0
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0,1]
[NeMo I 2024-05-13 16:53:27 nlp_overrides:150] Configuring DDP for model parallelism.
[NeMo I 2024-05-13 16:53:27 modelPT:728] Optimizer config = FusedAdam (
Parameter Group 0
betas: [0.9, 0.999]
bias_correction: True
eps: 1e-08
lr: 0.001
weight_decay: 0.1
Parameter Group 1
betas: [0.9, 0.999]
bias_correction: True
eps: 1e-08
lr: 0.001
weight_decay: 0.0
)
[NeMo I 2024-05-13 16:53:27 lr_scheduler:910] Scheduler "<nemo.core.optim.lr_scheduler.CosineAnnealing object at 0x7fed7615f220>"
will be used during training (effective maximum steps = 200) -
Parameters :
(warmup_steps: 2.0
constant_steps: 10.0
max_steps: 200
min_lr: 2.0e-05
)
| Name | Type | Params
----------------------------------------------------------------------------
0 | model | BertModel | 10.3 M
1 | model.language_model | TransformerLanguageModel | 10.2 M
2 | model.language_model.embedding | Embedding | 7.0 M
3 | model.language_model.encoder | ParallelTransformer | 3.2 M
4 | model.lm_head | BertLMHead | 91.8 K
5 | model.lm_head.dense | Linear | 65.8 K
6 | model.lm_head.layernorm | MixedFusedLayerNorm | 512
----------------------------------------------------------------------------
10.3 M Trainable params
0 Non-trainable params
10.3 M Total params
41.200 Total estimated model params size (MB)
Sanity Checking: 0it [00:00, ?it/s][NeMo W 2024-05-13 16:53:27 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/trainer/connectors/data_connector.py:438: PossibleUserWarning: The dataloader, val_dataloader, does not have many workers which may be a bottleneck. Consider increasing the value of the `num_workers` argument` (try 64 which is the number of cpus on this machine) in the `DataLoader` init to improve performance.
rank_zero_warn(
[NeMo W 2024-05-13 16:53:27 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/loops/utilities.py:148: UserWarning: Found `dataloader_iter` argument in the `validation_step`. Note that the support for this signature is experimental and the behavior is subject to change.
rank_zero_warn(
Sanity Checking DataLoader 0: 0%| | 0/2 [00:00<?, ?it/s][NeMo W 2024-05-13 16:53:27 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/modules/common/megatron/fused_bias_dropout_add.py:70: UserWarning: nvfuser integration in TorchScript is deprecated. (Triggered internally at /opt/pytorch/pytorch/torch/csrc/jit/codegen/cuda/interface.cpp:235.)
return bias_dropout_add_fused_inference_(*args)
Sanity Checking DataLoader 0: : 3it [00:01, 2.18it/s] [NeMo W 2024-05-13 16:53:28 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/trainer/connectors/logger_connector/result.py:433: PossibleUserWarning: It is recommended to use `self.log('val_loss', ..., sync_dist=True)` when logging on epoch level in distributed setting to accumulate the metric across devices.
warning_cache.warn(
[NeMo W 2024-05-13 16:53:28 nemo_logging:349] /usr/lib/python3.10/multiprocessing/popen_fork.py:66: RuntimeWarning: os.fork() was called. os.fork() is incompatible with multithreaded code, and JAX is multithreaded, so this will likely lead to a deadlock.
self.pid = os.fork()
[NeMo W 2024-05-13 16:53:29 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/loops/utilities.py:148: UserWarning: Found `dataloader_iter` argument in the `training_step`. Note that the support for this signature is experimental and the behavior is subject to change.
rank_zero_warn(
Epoch 0: 0%| | 0/200 [00:00<?, ?it/s][NeMo W 2024-05-13 16:53:30 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/trainer/connectors/logger_connector/result.py:212: UserWarning: You called `self.log('global_step', ...)` in your `training_step` but the value needs to be floating point. Converting it to torch.float32.
warning_cache.warn(
[NeMo W 2024-05-13 16:53:30 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/pytorch_lightning/trainer/connectors/logger_connector/result.py:212: UserWarning: You called `self.log('consumed_samples', ...)` in your `training_step` but the value needs to be floating point. Converting it to torch.float32.
warning_cache.warn(
Epoch 0: 50%|▌| 100/200 [00:30<00:30, 3.24it/s, v_num=3-24, reduced_train_loss
Validation: 0it [00:00, ?it/s]
Validation: 0%| | 0/8 [00:00<?, ?it/s]
Validation DataLoader 0: 0%| | 0/8 [00:00<?, ?it/s]
Validation DataLoader 0: 12%|██▌ | 1/8 [00:00<00:00, 7.28it/s]
Validation DataLoader 0: 25%|█████ | 2/8 [00:00<00:00, 7.40it/s]
Validation DataLoader 0: 38%|███████▌ | 3/8 [00:00<00:00, 7.57it/s]
Validation DataLoader 0: 50%|██████████ | 4/8 [00:00<00:00, 7.52it/s]
Validation DataLoader 0: 62%|████████████▌ | 5/8 [00:00<00:00, 7.50it/s]
Validation DataLoader 0: 75%|███████████████ | 6/8 [00:00<00:00, 7.58it/s]
Validation DataLoader 0: 88%|█████████████████▌ | 7/8 [00:00<00:00, 7.57it/s]
Validation DataLoader 0: 100%|████████████████████| 8/8 [00:01<00:00, 7.57it/s]
Epoch 0: 50%|▌| 100/200 [00:31<00:31, 3.13it/s, v_num=3-24, reduced_train_loss
Epoch 0, global step 100: 'val_loss' reached 9.03883 (best 9.03883), saving model to '/workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer--val_loss=9.04-step=100-consumed_samples=800.0.ckpt' as top 1
[NeMo I 2024-05-13 16:54:01 nemo_model_checkpoint:183] New .nemo model saved to: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
[NeMo I 2024-05-13 16:54:01 nemo_model_checkpoint:183] New .nemo model saved to: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
Epoch 0: 100%|█| 200/200 [01:02<00:00, 3.22it/s, v_num=3-24, reduced_train_loss
Validation: 0it [00:00, ?it/s]
Validation: 0%| | 0/8 [00:00<?, ?it/s]
Validation DataLoader 0: 0%| | 0/8 [00:00<?, ?it/s]
Validation DataLoader 0: 12%|██▌ | 1/8 [00:00<00:00, 7.58it/s]
Validation DataLoader 0: 25%|█████ | 2/8 [00:00<00:00, 7.47it/s]
Validation DataLoader 0: 38%|███████▌ | 3/8 [00:00<00:00, 7.49it/s]
Validation DataLoader 0: 50%|██████████ | 4/8 [00:00<00:00, 7.53it/s]
Validation DataLoader 0: 62%|████████████▌ | 5/8 [00:00<00:00, 7.51it/s]
Validation DataLoader 0: 75%|███████████████ | 6/8 [00:00<00:00, 7.51it/s]
Validation DataLoader 0: 88%|█████████████████▌ | 7/8 [00:00<00:00, 7.57it/s]
Validation DataLoader 0: 100%|████████████████████| 8/8 [00:01<00:00, 7.55it/s]
Epoch 0: 100%|█| 200/200 [01:03<00:00, 3.16it/s, v_num=3-24, reduced_train_loss
Epoch 0, global step 200: 'val_loss' reached 9.00171 (best 9.00171), saving model to '/workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer--val_loss=9.00-step=200-consumed_samples=1600.0.ckpt' as top 1
[NeMo I 2024-05-13 16:54:32 nemo_model_checkpoint:183] New .nemo model saved to: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
[NeMo I 2024-05-13 16:54:32 nlp_overrides:412] Removing checkpoint: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer--val_loss=9.04-step=100-consumed_samples=800.0.ckpt
[NeMo I 2024-05-13 16:54:32 nemo_model_checkpoint:183] New .nemo model saved to: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
[NeMo I 2024-05-13 16:54:33 nlp_overrides:412] Removing checkpoint: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer--val_loss=9.04-step=100-consumed_samples=800.0-last.ckpt
Epoch 0: 100%|█| 200/200 [01:04<00:00, 3.12it/s, v_num=3-24, reduced_train_loss`Trainer.fit` stopped: `max_steps=200` reached.
Epoch 0: 100%|█| 200/200 [01:04<00:00, 3.12it/s, v_num=3-24, reduced_train_loss
[NeMo I 2024-05-13 16:54:33 pretrain:49] *************** Finish Training ************
wandb: Waiting for W&B process to finish... (success).
wandb:
wandb: Run history:
wandb: consumed_samples ▁█
wandb: epoch ▁▁▁▁
wandb: global_step ▁█
wandb: grad_norm ▁█
wandb: lr █▁
wandb: reduced_train_loss █▁
wandb: train_backward_timing in s █▁
wandb: train_step_timing in s █▁
wandb: trainer/global_step ▄▁▁▁▁▁▁▁▁▁▄█▁▁▁▁▁▁▂▂▂█
wandb: val_loss █▁
wandb: validation_step_timing in s ██▇██▇██▁██████▇█▁
wandb:
wandb: Run summary:
wandb: consumed_samples 1600.0
wandb: epoch 0
wandb: global_step 199.0
wandb: grad_norm 0.73804
wandb: lr 2e-05
wandb: reduced_train_loss 8.66306
wandb: train_backward_timing in s 3e-05
wandb: train_step_timing in s 0.28713
wandb: trainer/global_step 199
wandb: val_loss 9.00171
wandb: validation_step_timing in s 0.00013
wandb:
wandb: You can sync this run to the cloud by running:
wandb: wandb sync /workspace/bionemo/data/singlecell_tutorial/inference_output/wandb/offline-run-20240513_165325-2024-05-13_16-53-24
wandb: Find logs at: /workspace/bionemo/data/singlecell_tutorial/inference_output/wandb/offline-run-20240513_165325-2024-05-13_16-53-24/logs
Running inference.#
We can see from the above training job that the model was trained for a short number of steps. Note the end of the log file the experiment manager leaves a message about where the resulting .nemo file is written. This file is used for finetuning, inference, or training from an existing set of model weights. See the example produced below from our run:
[NeMo I 2024-04-26 22:02:36 nemo_model_checkpoint:183] New .nemo model saved to: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-33-16/checkpoints/geneformer.nemo
[NeMo I 2024-04-26 22:02:36 nlp_overrides:412] Removing checkpoint: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-33-16/checkpoints/geneformer--val_loss=8.70-step=100-consumed_samples=800.0-last.ckpt
Epoch 0: 100%|█| 200/200 [00:27<00:00, 7.17it/s, v_num=2-05, reduced_train_loss`Trainer.fit` stopped: `max_steps=200` reached.
Epoch 0: 100%|█| 200/200 [00:27<00:00, 7.17it/s, v_num=2-05, reduced_train_loss
We will take the .nemo file logged:
/workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-04-26_22-02-05/checkpoints/geneformer.nemo
and use this for inference.
pretrained_nemo_file = '/workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo'
# Run inference on test
!python /workspace/bionemo/bionemo/model/infer.py \
--config-dir /workspace/bionemo/examples/singlecell/geneformer/conf \
--config-name infer \
++model.downstream_task.restore_from_path={pretrained_nemo_file} \
++model.data.dataset_path={test_tutorial_processed_dir} \
++exp_manager.exp_dir={tutorial_output_dir} \
++model.data.output_fname={tutorial_output_inference_pickle}
[NeMo W 2024-05-13 16:55:27 nemo_logging:349] /usr/lib/python3.10/multiprocessing/popen_fork.py:66: RuntimeWarning: os.fork() was called. os.fork() is incompatible with multithreaded code, and JAX is multithreaded, so this will likely lead to a deadlock.
self.pid = os.fork()
[NeMo I 2024-05-13 16:55:28 megatron_hiddens:110] Registered hidden transform sampled_var_cond_gaussian at bionemo.model.core.hiddens_support.SampledVarGaussianHiddenTransform
[NeMo I 2024-05-13 16:55:28 megatron_hiddens:110] Registered hidden transform interp_var_cond_gaussian at bionemo.model.core.hiddens_support.InterpVarGaussianHiddenTransform
[NeMo W 2024-05-13 16:55:28 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/hydra/_internal/hydra.py:119: UserWarning: Future Hydra versions will no longer change working directory at job runtime by default.
See https://hydra.cc/docs/1.2/upgrades/1.1_to_1.2/changes_to_job_working_dir/ for more information.
ret = run_job(
[NeMo I 2024-05-13 16:55:28 loading:31]
************** Experiment configuration ***********
[NeMo I 2024-05-13 16:55:28 loading:32]
name: geneformer_inference
desc: Minimum configuration for initializing a Geneformer model for inference.
trainer:
precision: bf16-mixed
devices: 1
num_nodes: 1
accelerator: gpu
logger: false
exp_manager:
explicit_log_dir: null
exp_dir: /workspace/bionemo/data/singlecell_tutorial/inference_output
name: ${name}
create_checkpoint_callback: false
model:
micro_batch_size: ${model.data.batch_size}
downstream_task:
restore_from_path: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
outputs:
- embeddings
- hiddens
data:
num_workers: 4
batch_size: 128
dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
output_fname: /workspace/bionemo/data/singlecell_tutorial/inference_output/human_covid19_bcells_from_scratch.pkl
index_mapping_dir: null
data_fields_map:
sequence: sequence
id: id
data_impl: geneformer
data_impl_kwargs:
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
dynamic_padding: true
post_process: false
inference_output_everything: false
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
infer_target: bionemo.model.singlecell.geneformer.infer.GeneformerInference
formatters:
simple:
format: '[%(asctime)s][%(name)s][%(levelname)s] - %(message)s'
handlers:
console:
class: logging.StreamHandler
formatter: simple
stream: ext://sys.stdout
file:
class: logging.FileHandler
formatter: simple
filename: /logs/inference.log
root:
level: INFO
handlers:
- console
disable_existing_loggers: false
[NeMo I 2024-05-13 16:55:28 utils:333] Restoring model from /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
[NeMo I 2024-05-13 16:55:28 utils:337] Loading model class: bionemo.model.singlecell.geneformer.model.GeneformerModel
GPU available: True (cuda), used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
HPU available: False, using: 0 HPUs
[NeMo I 2024-05-13 16:55:29 exp_manager:394] Experiments will be logged at /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer_inference/2024-05-13_16-55-29
[NeMo I 2024-05-13 16:55:29 exp_manager:835] TensorboardLogger has been set up
[NeMo I 2024-05-13 16:55:29 utils:306]
************** Trainer configuration ***********
[NeMo I 2024-05-13 16:55:29 utils:307]
name: geneformer_inference
desc: Minimum configuration for initializing a Geneformer model for inference.
trainer:
precision: bf16-mixed
devices: 1
num_nodes: 1
accelerator: gpu
logger: false
accumulate_grad_batches: 1
exp_manager:
explicit_log_dir: null
exp_dir: /workspace/bionemo/data/singlecell_tutorial/inference_output
name: ${name}
create_checkpoint_callback: false
model:
tokenizer:
vocab_file: nemo:92fcea8a75d2427ea07b5094b6d6cb20_geneformer.vocab
micro_batch_size: ${model.data.batch_size}
activation: relu
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
use_flash_attention: true
seq_length: 2048
encoder_seq_length: 2048
max_position_embeddings: 2048
num_layers: 6
hidden_size: 256
ffn_hidden_size: 512
num_attention_heads: 4
init_method_std: 0.02
hidden_dropout: 0.02
attention_dropout: 0.02
kv_channels: null
apply_query_key_layer_scaling: true
layernorm_epsilon: 1.0e-12
make_vocab_size_divisible_by: 128
pre_process: true
post_process: false
bert_binary_head: false
resume_from_checkpoint: null
masked_softmax_fusion: true
native_amp_init_scale: 4294967296
native_amp_growth_interval: 1000
fp32_residual_connection: true
fp16_lm_cross_entropy: false
seed: 1234
use_cpu_initialization: false
onnx_safe: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
data:
data_impl: geneformer
probabilistic_dirichlet_sampling_train: false
train_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/train
val_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/val
test_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
dataset: /
data_prefix: ''
shuffle: true
medians_file: nemo:b94a7da5807840419b10b3bf4c6126fe_medians.json
index_mapping_dir: null
skip_warmup: true
index_mapping_type: memmap
num_workers: 4
dataloader_type: single
seq_length: 2048
seed: 1234
dynamic_padding: true
micro_batch_size: 8
output_fname: /workspace/bionemo/data/singlecell_tutorial/inference_output/human_covid19_bcells_from_scratch.pkl
batch_size: 128
data_fields_map:
sequence: sequence
id: id
data_impl_kwargs:
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
optim:
name: fused_adam
lr: 0.001
weight_decay: 0.1
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: 2.0
constant_steps: 10.0
max_steps: 200
min_lr: 2.0e-05
global_batch_size: 128
precision: bf16-mixed
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
nemo_version: 1.22.0
downstream_task:
restore_from_path: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
outputs:
- embeddings
- hiddens
inference_output_everything: false
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
infer_target: bionemo.model.singlecell.geneformer.infer.GeneformerInference
formatters:
simple:
format: '[%(asctime)s][%(name)s][%(levelname)s] - %(message)s'
handlers:
console:
class: logging.StreamHandler
formatter: simple
stream: ext://sys.stdout
file:
class: logging.FileHandler
formatter: simple
filename: /logs/inference.log
root:
level: INFO
handlers:
- console
disable_existing_loggers: false
[NeMo W 2024-05-13 16:55:29 modelPT:251] You tried to register an artifact under config key=tokenizer.vocab_file but an artifact for it has already been registered.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: pipeline_model_parallel_split_rank in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo I 2024-05-13 16:55:29 megatron_init:234] Rank 0 has data parallel group: [0]
[NeMo I 2024-05-13 16:55:29 megatron_init:237] All data parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:55:29 megatron_init:238] Ranks 0 has data parallel rank: 0
[NeMo I 2024-05-13 16:55:29 megatron_init:246] Rank 0 has model parallel group: [0]
[NeMo I 2024-05-13 16:55:29 megatron_init:247] All model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:55:29 megatron_init:257] Rank 0 has tensor model parallel group: [0]
[NeMo I 2024-05-13 16:55:29 megatron_init:261] All tensor model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:55:29 megatron_init:262] Rank 0 has tensor model parallel rank: 0
[NeMo I 2024-05-13 16:55:29 megatron_init:276] Rank 0 has pipeline model parallel group: [0]
[NeMo I 2024-05-13 16:55:29 megatron_init:288] Rank 0 has embedding group: [0]
[NeMo I 2024-05-13 16:55:29 megatron_init:294] All pipeline model parallel group ranks: [[0]]
[NeMo I 2024-05-13 16:55:29 megatron_init:295] Rank 0 has pipeline model parallel rank 0
[NeMo I 2024-05-13 16:55:29 megatron_init:296] All embedding group ranks: [[0]]
[NeMo I 2024-05-13 16:55:29 megatron_init:297] Rank 0 has embedding rank: 0
24-05-13 16:55:29 - PID:294681 - rank:(0, 0, 0, 0) - microbatches.py:39 - INFO - setting number of micro-batches to constant 1
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: context_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: virtual_pipeline_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: sequence_parallel in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: expert_model_parallel_size in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: gradient_accumulation_fusion in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_overlap in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_ag in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_split_rs in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_wgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: tp_comm_bulk_dgrad in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: finalize_model_grads_func in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: overlap_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: batch_p2p_comm in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: pipeline_model_parallel_split_rank in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 megatron_base_model:821] The model: GeneformerModel() does not have field.name: barrier_with_L1_time in its cfg. Add this key to cfg or config_mapping to make to make it configurable.
[NeMo W 2024-05-13 16:55:29 modelPT:251] You tried to register an artifact under config key=tokenizer.vocab_file but an artifact for it has already been registered.
[NeMo I 2024-05-13 16:55:29 megatron_base_model:315] Padded vocab_size: 25472, original vocab_size: 25429, dummy tokens: 43.
[NeMo I 2024-05-13 16:55:30 nlp_overrides:752] Model GeneformerModel was successfully restored from /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo.
[NeMo I 2024-05-13 16:55:30 utils:471] DDP is not initialized. Initializing...
Initializing distributed: GLOBAL_RANK: 0, MEMBER: 1/1
----------------------------------------------------------------------------------------------------
distributed_backend=nccl
All distributed processes registered. Starting with 1 processes
----------------------------------------------------------------------------------------------------
[NeMo W 2024-05-13 16:55:30 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/apex/transformer/pipeline_parallel/utils.py:81: UserWarning: This function is only for unittest
warnings.warn("This function is only for unittest")
[NeMo W 2024-05-13 16:55:30 nemo_logging:349] /usr/local/lib/python3.10/dist-packages/nemo/collections/nlp/modules/common/megatron/fused_bias_dropout_add.py:70: UserWarning: nvfuser integration in TorchScript is deprecated. (Triggered internally at /opt/pytorch/pytorch/torch/csrc/jit/codegen/cuda/interface.cpp:235.)
return bias_dropout_add_fused_inference_(*args)
[NeMo I 2024-05-13 16:55:31 loading:43]
************** Restored model configuration ***********
[NeMo I 2024-05-13 16:55:31 loading:44]
tokenizer:
vocab_file: /tmp/tmp13zi86ni/92fcea8a75d2427ea07b5094b6d6cb20_geneformer.vocab
micro_batch_size: 128
activation: relu
tensor_model_parallel_size: 1
pipeline_model_parallel_size: 1
use_flash_attention: true
seq_length: 2048
encoder_seq_length: 2048
max_position_embeddings: 2048
num_layers: 6
hidden_size: 256
ffn_hidden_size: 512
num_attention_heads: 4
init_method_std: 0.02
hidden_dropout: 0.02
attention_dropout: 0.02
kv_channels: null
apply_query_key_layer_scaling: true
layernorm_epsilon: 1.0e-12
make_vocab_size_divisible_by: 128
pre_process: true
post_process: false
bert_binary_head: false
resume_from_checkpoint: null
masked_softmax_fusion: true
native_amp_init_scale: 4294967296
native_amp_growth_interval: 1000
fp32_residual_connection: true
fp16_lm_cross_entropy: false
seed: 1234
use_cpu_initialization: false
onnx_safe: false
activations_checkpoint_method: null
activations_checkpoint_num_layers: 1
data:
data_impl: geneformer
probabilistic_dirichlet_sampling_train: false
train_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/train
val_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/val
test_dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
dataset_path: /workspace/bionemo/data/singlecell_tutorial/processed_data/test
dataset: /
data_prefix: ''
shuffle: true
medians_file: nemo:b94a7da5807840419b10b3bf4c6126fe_medians.json
index_mapping_dir: null
skip_warmup: true
index_mapping_type: memmap
num_workers: 4
dataloader_type: single
seq_length: 2048
seed: 1234
dynamic_padding: true
micro_batch_size: 8
output_fname: /workspace/bionemo/data/singlecell_tutorial/inference_output/human_covid19_bcells_from_scratch.pkl
batch_size: 128
data_fields_map:
sequence: sequence
id: id
data_impl_kwargs:
csv_fields_mmap:
newline_int: 10
header_lines: 1
workers: null
sort_dataset_paths: false
data_sep: ','
data_fields:
id: 0
sequence: 1
fasta_fields_mmap:
data_fields:
id: 0
sequence: 1
optim:
name: fused_adam
lr: 0.001
weight_decay: 0.1
betas:
- 0.9
- 0.999
sched:
name: CosineAnnealing
warmup_steps: 2.0
constant_steps: 10.0
max_steps: 200
min_lr: 2.0e-05
global_batch_size: 128
precision: bf16-mixed
target: bionemo.model.singlecell.geneformer.model.GeneformerModel
nemo_version: 1.22.0
downstream_task:
restore_from_path: /workspace/bionemo/data/singlecell_tutorial/inference_output/geneformer/2024-05-13_16-53-24/checkpoints/geneformer.nemo
outputs:
- embeddings
- hiddens
inference_output_everything: false
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0,1]
[NeMo W 2024-05-13 16:55:31 nemo_logging:349] /usr/lib/python3.10/multiprocessing/popen_fork.py:66: RuntimeWarning: os.fork() was called. os.fork() is incompatible with multithreaded code, and JAX is multithreaded, so this will likely lead to a deadlock.
self.pid = os.fork()
Predicting DataLoader 0: 100%|████████████████████| 2/2 [00:00<00:00, 2.14it/s]
[NeMo I 2024-05-13 16:55:32 run_inference:50] Collecting results from all GPUs...
[NeMo I 2024-05-13 16:55:32 infer:73] Saving 232 samples to /workspace/bionemo/data/singlecell_tutorial/inference_output/human_covid19_bcells_from_scratch.pkl
Load inference result and cluster with UMAP.#
Now we will inspect our result. First, we expect there to be one prediction for each cell, we can compare the shape of the anndata object to the predictions produced by our model. After this, we can simply pass our embeddings into umap, and view the result! In this case its a very poorly trained model with very few cells, so keep expectations low!
The inference_results pickle file contains one set of hiddens and embeddings for each cell. The hiddens contain the embedding per-token, whereas the embeddings contain the mean embedding for all gene tokens with special tokens (CLS, MASK, etc) removed.
import pickle
with open(tutorial_output_inference_pickle, 'rb') as inference_handle:
inference_results = pickle.load(inference_handle)
len(inference_results), adata.shape, inference_results[0].keys()
(232, (2313, 60664), dict_keys(['embeddings']))
inference_results[0]['embeddings'].shape
(256,)
import umap
reducer = umap.UMAP()
embedding = reducer.fit_transform([x['embeddings'] for x in inference_results])
embedding.shape
(232, 2)
assert adata_test.obs.shape[0] == len(inference_results)
from matplotlib import pyplot as plt
results = adata_test.obs.copy()
results['x'] = embedding[:, 0]
results['y'] = embedding[:, 1]
covariates = ["assay", "development_stage", "dataset_id", "sex"]
fig, axes = plt.subplots(nrows=2, ncols=2, sharex=True, sharey=True, figsize=(10,10))
for ax,covar in zip(axes.flat, covariates):
for cov, cov_df in results.groupby(covar):
ax.scatter(
cov_df.x,
cov_df.y,
s=3,
alpha=0.75,
label=cov,
)
if len(results[covar].unique()) < 8:
ax.legend()
ax.set_title(f"Embeddings by {covar}")
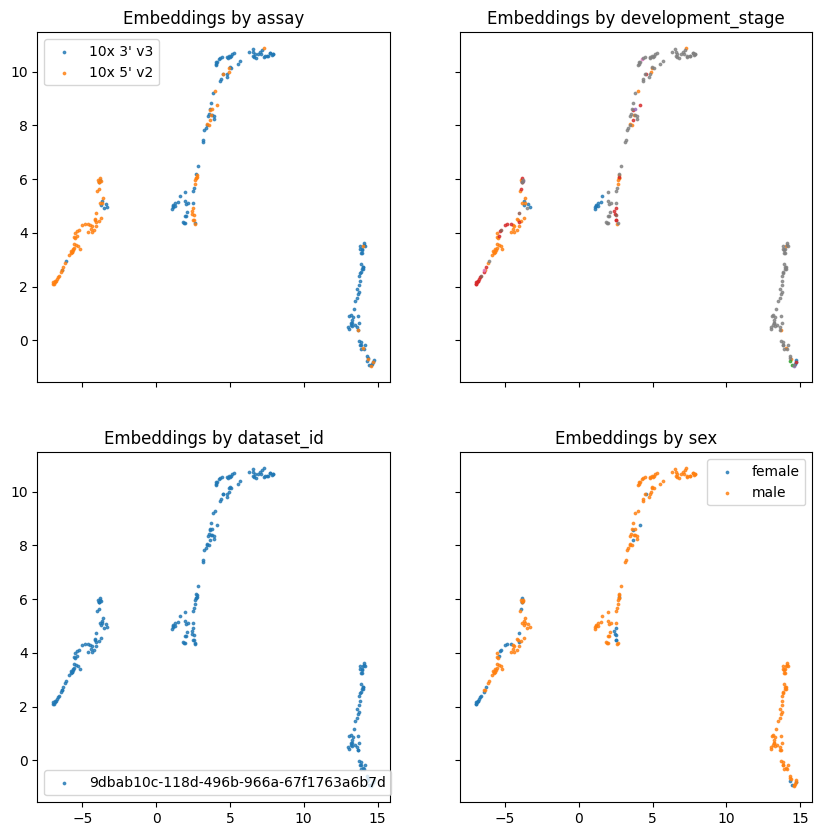
adata_test.obs.columns
Index(['soma_joinid', 'dataset_id', 'assay', 'assay_ontology_term_id',
'cell_type', 'cell_type_ontology_term_id', 'development_stage',
'development_stage_ontology_term_id', 'disease',
'disease_ontology_term_id', 'donor_id', 'is_primary_data',
'self_reported_ethnicity', 'self_reported_ethnicity_ontology_term_id',
'sex', 'sex_ontology_term_id', 'suspension_type', 'tissue',
'tissue_ontology_term_id', 'tissue_general',
'tissue_general_ontology_term_id', 'raw_sum', 'nnz', 'raw_mean_nnz',
'raw_variance_nnz', 'n_measured_vars'],
dtype='object')